Clear Sky Science · fr
Formation stéréosélective de liaisons C(sp3)–C vicinales via une 1,2-difonctionnalisation métallophotoredox d’alcènes internes
Construire des molécules complexes avec la lumière
Les chimistes doivent souvent assembler de petits fragments carbonés en architectures tridimensionnelles complexes pour les médicaments et les matériaux. Le faire rapidement, proprement et de façon contrôlée est étonnamment difficile, surtout lorsque deux nouvelles liaisons carbone–carbone doivent se former côte à côte en une seule étape. Cette étude présente une méthode photodirigée qui utilise des catalyseurs au nickel pour assembler de tels cadres carbonés densément substitués avec un niveau de précision jusque-là difficile à atteindre, ouvrant des voies plus rapides vers des molécules de type médicament.
Pourquoi les liaisons carbonées voisines sont difficiles
Beaucoup de médicaments modernes fonctionnent mieux lorsque leurs atomes de carbone sont arrangés selon un motif tridimensionnel précis, un peu comme une clé taillée pour une serrure. Un motif particulièrement précieux implique deux atomes de carbone voisins qui deviennent chacun un nouveau point de ramification, transformant une double liaison carbone–carbone plate en une structure saturée et compacte. Les alcènes internes — doubles liaisons situées à l’intérieur d’une molécule plutôt qu’en bout de chaîne — sont des points de départ attrayants pour cela, mais ils posent deux problèmes. Leur encombrement ralenti la première addition radicalaire, et une fois le premier fragment fixé, deux fragments carbonés de réactivité similaire peuvent entrer en compétition de façon incontrôlée, rendant difficile de décider quel fragment se lie où et sur quelle face de la molécule.
Utiliser la lumière et le nickel comme une équipe coordonnée
Les chercheurs ont conçu une plate-forme « métallophotoredox », dans laquelle la lumière visible excite un photocatalyseur ou une source radicalaire spéciale tandis qu’un complexe de nickel guide la formation des liaisons. Ensemble, ces catalyseurs génèrent des radicaux carbonés de courte durée de vie qui s’additionnent aux alcènes internes, puis canalisent l’intermédiaire résultant vers une étape de couplage catalysée par le nickel qui fixe un second fragment carboné. En choisissant différents ligands — les petites molécules liées au nickel — ils peuvent basculer entre deux modes utiles. Avec un ligand terpyridine et deux partenaires alkyles, le système réalise une 1,2-dialkylation, installant deux ramifications carbonées en disposition « anti ». Avec un ligand chiral biimidazole et un alkyle plus un fragment aryle, il effectue une 1,2-alkylarylation, encore en anti, mais en outre en sélectionnant une seule forme image miroir avec une grande précision.
Ce que les nouvelles réactions peuvent construire
Sous une irradiation douce par lumière bleue, le protocole de dialkylation convertit une variété d’alcènes internes électroattracteurs cycliques et acycliques en produits portant deux nouvelles liaisons C(sp3)–C voisines, souvent avec de bons rendements et un excellent contrôle de la face sur laquelle chaque fragment se place. La méthode tolère de nombreux groupes fonctionnels et fonctionne même sur des produits naturels complexes et des sondes fluorescentes, permettant aux chimistes d’ajouter des « poignées » riches en sp3 tard dans une synthèse. La variante alkylarylation, utilisant un catalyseur chiral nickel–biimidazole et un photocatalyseur séparé, atteint à la fois une forte diastéréosélectivité et une haute énantiosélectivité. Elle transforme des coumarines, quinolinones et cycles apparentés en lactones β-aryl-α-alkyl et en échafaudages similaires contenant deux centres stéréogènes adjacents, des structures recherchées en chimie médicinale.
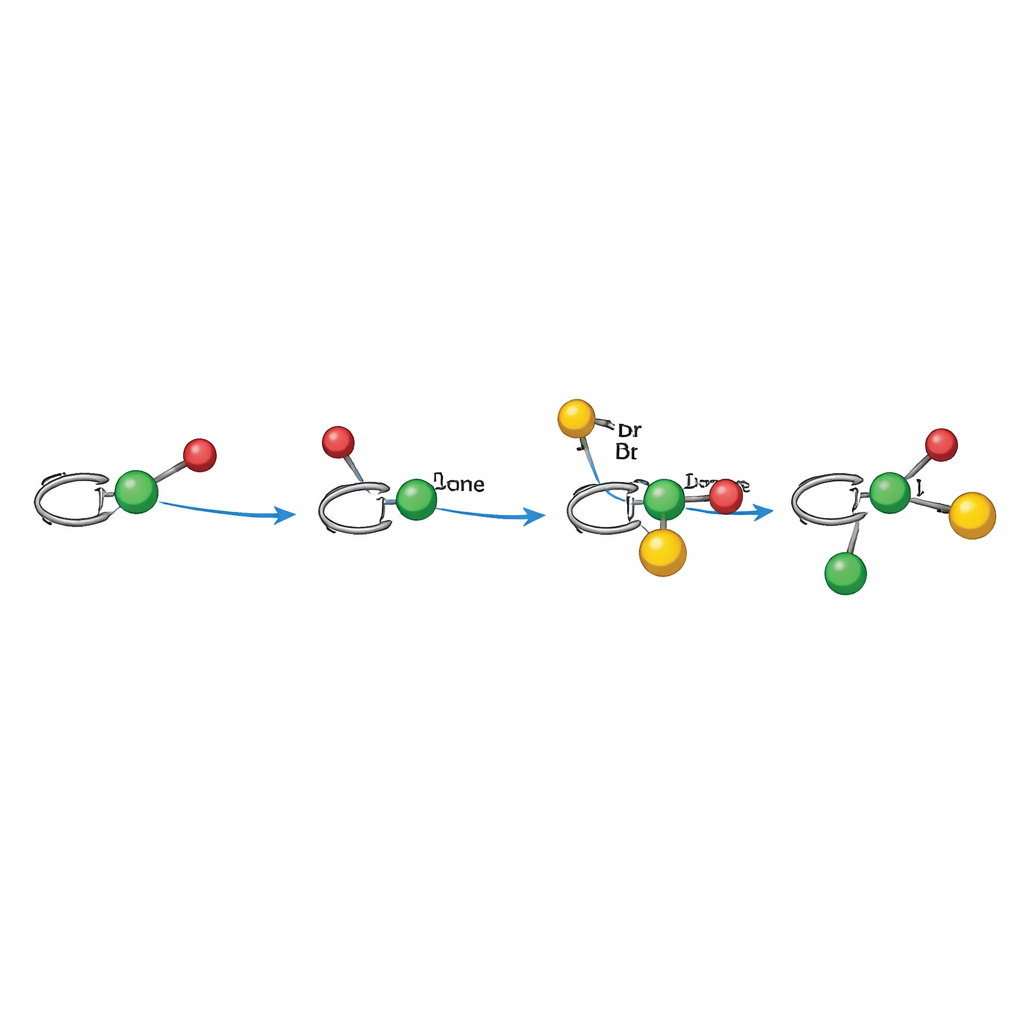
Comment fonctionne le mécanisme photodirigé
Des expériences mécanistiques soutiennent une voie radicalaire minutieusement orchestrée par le nickel. En mode alkylarylation, la lumière active d’abord le photocatalyseur, qui génère un radical alkyle à partir d’un ester à activité rédox. Ce radical s’ajoute à l’alcène, formant un radical benzylique qui est lié — directement ou indirectement — à un centre nickel chiral ayant déjà engagé l’halogénoaryle. Le complexe de nickel capture alors le radical benzylique et forge la nouvelle liaison carbone–carbone, libérant le produit et régénérant le catalyseur par transfert d’électron. En mode dialkylation, un ester de Hantzsch sert doublement de source radicalaire et de photosensibilisateur interne, réduisant directement le nickel et produisant un radical alkyle sous lumière. Dans les deux cas, la géométrie du ligand autour du nickel dirige la face de l’alcène attaquée et la manière dont les liaisons finales se forment, expliquant le fort contrôle de l’arrangement tridimensionnel.
Ce que cela signifie pour les molécules futures
En transformant une double liaison interne récalcitrante en une porte d’entrée vers deux ramifications carbonées précisément positionnées, ce travail offre aux chimistes un raccourci puissant vers des cadres tridimensionnels complexes. Les deux protocoles — l’un pour des fragments purement alkyles et l’autre pour alkyle plus aryle — opèrent dans des conditions douces et réductrices et tolèrent de nombreux groupes sensibles, ce qui les rend particulièrement adaptés aux modifications en phase finale de candidats-médicaments. Pour un non-spécialiste, l’idée principale est que l’utilisation conjointe de la lumière et du nickel fournit une chaîne d’assemblage finement réglée pour construire des voisinages carbonés encombrés auparavant difficiles d’accès, accélérant potentiellement la découverte et l’optimisation de nouveaux médicaments.
Citation: Zhang, Y., Long, T., Sun, Y. et al. Stereoselective vicinal C(sp³)–C bond formation via metallaphotoredox 1,2-difunctionalization of internal alkenes. Nat Commun 17, 3066 (2026). https://doi.org/10.1038/s41467-026-69838-7
Mots-clés: dicarbofonctionnalisation radicalaire, catalyse métallophotoredox, catalyse au nickel, alcènes internes, formation stéréosélective de liaisons C–C