Clear Sky Science · de
Stereoselektive vicinale C(sp³)–C-Bindungsbildung durch metallaphotoredox 1,2-Difunktionalisierung interner Alkene
Komplexe Moleküle mit Licht aufbauen
Chemiker müssen kleine Kohlenstofffragmente oft zu komplexen dreidimensionalen Formen zusammensetzen, etwa für Medikamente und Materialien. Das schnell, sauber und kontrolliert zu tun, ist überraschend schwierig — besonders wenn in einem einzigen Schritt zwei neue Kohlenstoff–Kohlenstoff-Bindungen nebeneinander entstehen sollen. Diese Studie stellt eine lichtgetriebene Methode vor, die Nickel-Katalysatoren nutzt, um derartige dicht gepackte Kohlenstoffgerüste mit einer Präzision zusammenzubauen, die bisher schwer erreichbar war, und eröffnet damit schnellere Wege zu medikamentenähnlichen Molekülen.
Warum benachbarte Kohlenstoffverknüpfungen schwierig sind
Viele moderne Wirkstoffe funktionieren am besten, wenn ihre Kohlenstoffatome eine bestimmte dreidimensionale Anordnung einnehmen, ähnlich wie ein Schlüssel, der auf ein bestimmtes Schloss zugeschnitten ist. Ein besonders wertvolles Muster sind zwei benachbarte Kohlenstoffatome, die jeweils neue Verzweigungspunkte werden und eine flache Doppelbindung in eine kompakte, gesättigte Struktur überführen. Interne Alkene — Doppelbindungen, die im Inneren eines Moleküls liegen statt am Ende — sind attraktive Ausgangspunkte dafür, stellen aber zwei Probleme. Ihre Sperrigkeit verlangsamt die erste radikalische Addition, und nachdem das erste neue Fragment angelagert ist, können zwei ähnlich reaktive Kohlenstofffragmente unkontrolliert konkurrieren, sodass schwer zu steuern ist, welches Fragment wo und an welcher Seite des Moleküls bindet.
Licht und Nickel als koordiniertes Team
Die Forscher entwarfen eine „metallaphotoredox“-Plattform, bei der sichtbares Licht einen Photokatalysator oder eine spezielle Radikalquelle anregt, während ein Nickelkomplex die Bindungsbildung lenkt. Gemeinsam erzeugen diese Katalysatoren kurzlebige Kohlenstoffradikale, die an interne Alkene addieren und das entstehende Zwischenprodukt in einen nickelbasierten Kupplungsschritt leiten, der ein zweites Kohlenstofffragment fixiert. Durch die Wahl verschiedener Liganden — der kleinen Moleküle, die an Nickel gebunden sind — lassen sich zwei nützliche Modi umschalten. Mit einem Terpyridin-Liganden und zwei Alkylpartnern führt das System eine 1,2-Dialkylierung durch und installiert zwei Kohlenstoffverzweigungen in einer „anti“-Anordnung. Mit einem chiralen Biimidazol-Liganden und je einem Alkyl- sowie einem Arylfragment findet eine 1,2-Alkylarylierungs-Reaktion statt, ebenfalls anti, die außerdem eine einzelne spiegelbildliche Form mit hoher Präzision auswählt.
Was die neuen Reaktionen aufbauen können
Unter milden Blau-Licht-Bestrahlungsbedingungen wandelt das Dialkylierungsprotokoll verschiedene cyclische und acyclische elektronenziehende interne Alkene in Produkte um, die zwei neue benachbarte C(sp³)–C-Bindungen tragen — oft mit hoher Ausbeute und ausgezeichneter Kontrolle darüber, auf welcher Seite der Doppelbindung die Fragmente liegen. Die Methode toleriert viele funktionelle Gruppen und funktioniert sogar an komplexen Naturstoffen und fluoreszenten Sonden, sodass Chemiker sp³-reiche „Griffe“ spät in einer Synthese anbringen können. Die Alkylarylierungs-Variante, die einen chiralen Nickel–Biimidazol-Katalysator und einen separaten Photokatalysator nutzt, erzielt sowohl hohe Diastereoselektivität als auch Enantioselektivität. Sie verwandelt Coumarine, Chinolinone und verwandte Ringe in β-Aryl-α-alkyl-Lactone und ähnliche Gerüste, die zwei benachbarte stereogene Zentren enthalten — Strukturen, die in der medizinischen Chemie geschätzt werden.
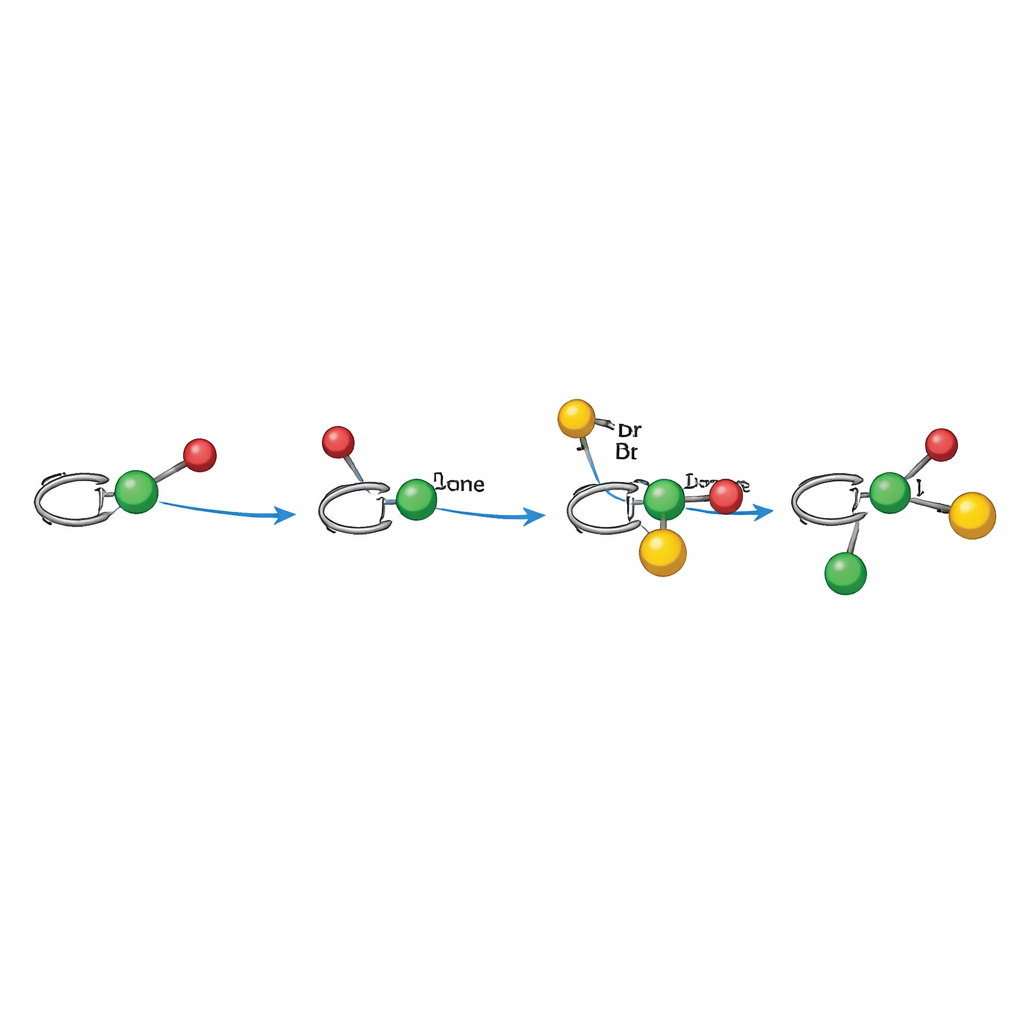
Wie der lichtgetriebene Mechanismus funktioniert
Mechanistische Experimente stützen einen radikalischen Pfad, der sorgfältig durch Nickel choreografiert wird. Im Alkylarylierungs-Modus aktiviert Licht zunächst den Photokatalysator, der aus einem redoxaktiven Ester ein Alkylradikal erzeugt. Dieses Radikal addiert an das Alken und bildet ein benzylicales Radikal, das direkt oder indirekt an ein chirales Nickelzentrum gebunden ist, das bereits mit dem Arylhalogen reagiert hat. Der Nickelkomplex fängt dann das benzylicale Radikal ein und schmiedet die neue Kohlenstoff–Kohlenstoff-Bindung, setzt das Produkt frei und regeneriert den Katalysator durch Elektronentransfer. Im Dialkylierungs-Modus dient ein Hantzsch-Ester gleichzeitig als Radikalquelle und als internes Photosensibilisator, reduziert Nickel direkt und erzeugt unter Lichteinfluss ein Alkylradikal. In beiden Fällen lenkt die Form des Liganden um das Nickel, welche Seite des Alkens angegriffen wird und wie die finalen Bindungen geschlossen werden — das erklärt die starke Kontrolle über die dreidimensionale Anordnung.
Was das für künftige Moleküle bedeutet
Indem eine hartnäckige interne Doppelbindung in ein Tor für zwei präzise platzierte Kohlenstoffverzweigungen verwandelt wird, bietet diese Arbeit Chemikern eine wirkungsvolle Abkürzung zu komplexen, dreidimensionalen Gerüsten. Die beiden Protokolle — eines für rein alkylische Fragmente und eines für Alkyl plus Aryl — arbeiten unter milden, reduzierenden Bedingungen und tolerieren viele empfindliche Gruppen, wodurch sie sich besonders für spätes Feintuning von Arzneimittelkandidaten eignen. Für Nichtfachleute lautet die Kernaussage: Die Kombination von Licht und Nickel bietet eine fein abgestimmte «Montagelinie» zum Aufbau dicht gepackter Kohlenstoffnachbarschaften, die zuvor schwer zugänglich waren, und kann so die Entdeckung und Optimierung neuer Wirkstoffe beschleunigen.
Zitation: Zhang, Y., Long, T., Sun, Y. et al. Stereoselective vicinal C(sp³)–C bond formation via metallaphotoredox 1,2-difunctionalization of internal alkenes. Nat Commun 17, 3066 (2026). https://doi.org/10.1038/s41467-026-69838-7
Schlüsselwörter: radikale Dicarbofunktionalisierung, metallaphotoredox-Katalyse, Nickel-Katalyse, interne Alkene, stereoselektive C–C-Bindungsbildung