Clear Sky Science · sv
SamplingDesign: RNA-design via kontinuerlig optimering med kopplade variabler och Monte Carlo-sampling
Att designa RNA som ett nytt verktyg för medicin
RNA är inte bara en passiv budbärare av genetisk information; det kan veckas till invecklade former som kontrollerar gener, katalyserar reaktioner och till och med utgör grunden för vacciner. Om forskare kunde pålitligt designa RNA-sekvenser som veckas till valda former skulle de kunna bygga skräddarsydda molekylära verktyg för medicin, från smartare vacciner till programmerbara genetiska strömbrytare. Denna artikel presenterar SamplingDesign, en ny beräkningsmetod som tar sig an den svåra uppgiften att designa RNA genom att blanda idéer från fysik, statistik och modern maskininlärning.
Varför det är så svårt att designa RNA-former
Att designa RNA är som att försöka välja en bokstavsstring så att den, när den vikt ihop sig, bildar precis rätt origamifigur — och nästan inget annat. För en kedja av längd n finns 4n möjliga sekvenser, så även måttliga längder exploderar till astronomiska mängder. Utöver det kan varje sekvens veckas till ett enormt antal alternativa former som konkurrerar med den önskade. En användbar design måste inte bara favorisera målformen som sitt lägsta-energitillstånd utan också göra konkurrerande former mycket mindre sannolika, så att rätt struktur dominerar i mängden av möjligheter. Traditionella sökmetoder justerar en eller några positioner åt gången och fastnar snabbt i detta labyrintiska alternativlandskap, särskilt för långa och komplexa RNA.
Ett nytt sätt att utforska möjligheter på en gång
I stället för att gå från en kandidatsekvens till nästa tänker SamplingDesign i termer av ett helt moln av möjligheter. Metoden börjar med en sannolikhetsfördelning utbredd över alla sekvenser som är kompatibla med målformen — det vill säga sekvenser vars parade positioner kan bilda verkliga kemiska baspar. Därefter använder metoden gradientbaserad optimering, en arbetsmotor inom maskininlärning, för att stadigt omforma denna fördelning så att sekvenser som sannolikt veckas väl till målstrukturen får högre sannolikhet medan dåliga kandidater tappar. Avgörande är att författarna inte optimerar en enskild poäng för en enstaka sekvens; de optimerar den genomsnittliga prestationen för alla sekvenser under den aktuella fördelningen, vilket uppmuntrar bred utforskning tidigt och finjustering senare.
Att fånga hur baser samverkar
Nyckeln i detta angreppssätt är ett mer realistiskt sätt att representera hur positioner längs RNA påverkar varandra. I stället för att behandla varje nukleotid som ett oberoende val, grupperar SamplingDesign vissa positioner i ”kopplade variabler”. För varje baspar delar de två partnerna en liten gemensam sannolikhetstabell över de sex kemiskt tillåtna parstyperna, vilket automatiskt utesluter ogiltiga kombinationer. Liknande koppling används för närliggande positioner som påverkar varandras energi, till exempel mismatch- och trimismatch-grupper runt slingor. Detta krymper designutrymmet till endast giltiga sekvenser och gör optimeringen smidigare, eftersom uppdateringar verkar direkt på meningsfulla baspars- och mismatch-val istället för på isolerade bokstäver.
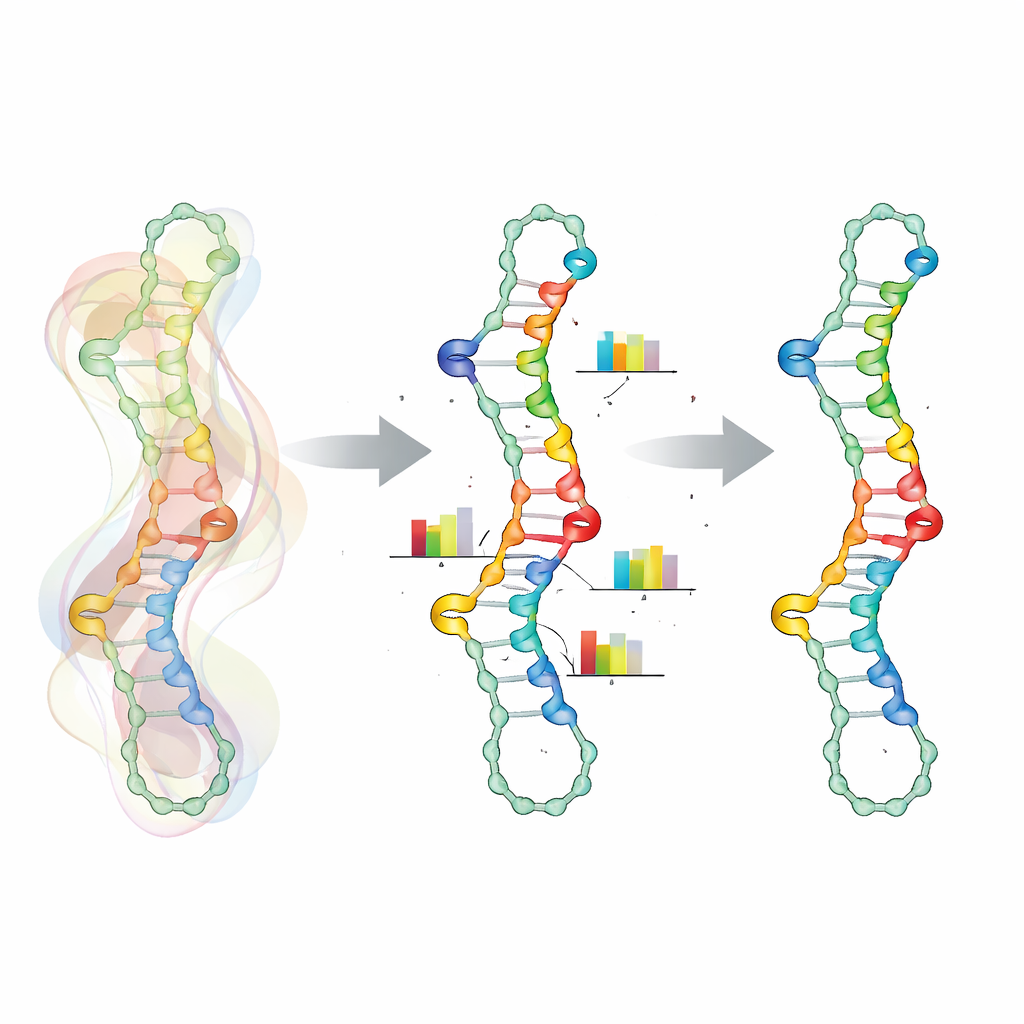
Låta slumpen styra smartare val
Eftersom det är omöjligt att beräkna exakta medelvärden över det enorma rummet av sekvenser och veckningar förlitar sig SamplingDesign på Monte Carlo-sampling. Vid varje steg drar metoden ett hanterbart antal sekvenser från den aktuella fördelningen, utvärderar hur väl var och en veckas enligt termodynamiska modeller och använder dessa prover för att uppskatta både det genomsnittliga objektivet (såsom sannolikheten för målstrukturen) och hur fördelningen ska justeras. Över många iterationer förskjuts sannolikhetsmassan mot bättre sekvenser och fördelningen blir skarpare. I stället för att enkelt välja den enskilt mest sannolika sekvensen i slutet håller metoden reda på alla prover den sett och väljer den som faktiskt presterar bäst enligt det valda måttet, vilket ger fördelarna av bred utforskning utan att tappa fokus.
Bättre än befintliga verktyg på svåra pussel
Författarna testade SamplingDesign på flera standardkollektioner av RNA-”pussel”, inklusive den vida använda Eterna100-benchmarken, som sträcker sig från enkla hårnålar till långa, intrikata former på upp till 400 nukleotider. I nästan alla mått som betraktar hela ensemblet av strukturer — särskilt Boltzmann-sannolikheten för målformen och ”ensemble defect”, som spårar hur ofta nukleotider felveckas — överträffade SamplingDesign toppmoderna designverktyg som förlitar sig på lokalsökning eller enklare kontinuerliga metoder. Fördelen var mest påtaglig för de längsta och svåraste pusslen, där traditionella algoritmer ofta fastnar i dåliga lösningar medan SamplingDesign fortsätter hitta sekvenser vars målformer framträder tydligt från konkurrenterna.
Vad detta betyder för framtida RN A-teknologier
Enkelt uttryckt visar detta arbete att om man betraktar RNA-design som en guidad utforskning av många sekvenser samtidigt, istället för ett steg-för-steg-redigeringsspel, kan man producera renare, mer tillförlitliga veckningar — särskilt för stora och utmanande mål. Genom att modellera hur baser interagerar i par och grupper, och genom att använda sampling för att navigera i ett annars ogenomträngligt landskap, erbjuder SamplingDesign en flexibel ram som kan optimera olika designmål. Författarna föreslår att det kan utvidgas för att skräddarsy budbärar-RNA för vacciner eller terapier och för att införliva experimentella begränsningar. När förbättrad beräkningsdesign möter laboratorietester kan sådana metoder hjälpa till att förvandla abstrakta RNA-blåkopior till praktiska molekylära verktyg för medicin.
Citering: Tang, W.Y., Dai, N., Zhou, T. et al. SamplingDesign: RNA design via continuous optimization with coupled variables and Monte-Carlo sampling. Nat Commun 17, 2950 (2026). https://doi.org/10.1038/s41467-025-67901-3
Nyckelord: RNA-design, invers vikning, Monte Carlo-sampling, kontinuerlig optimering, mRNA-terapeutika