Clear Sky Science · en
SamplingDesign: RNA design via continuous optimization with coupled variables and Monte-Carlo sampling
Designing RNA as a new tool for medicine
RNA is not just a passive messenger of genetic information; it can fold into intricate shapes that control genes, catalyze reactions, and even serve as the basis of vaccines. If scientists could reliably design RNA sequences that fold into chosen shapes, they could build custom molecular tools for medicine, from smarter vaccines to programmable genetic switches. This paper introduces SamplingDesign, a new computational method that tackles the daunting difficulty of RNA design by blending ideas from physics, statistics, and modern machine learning.
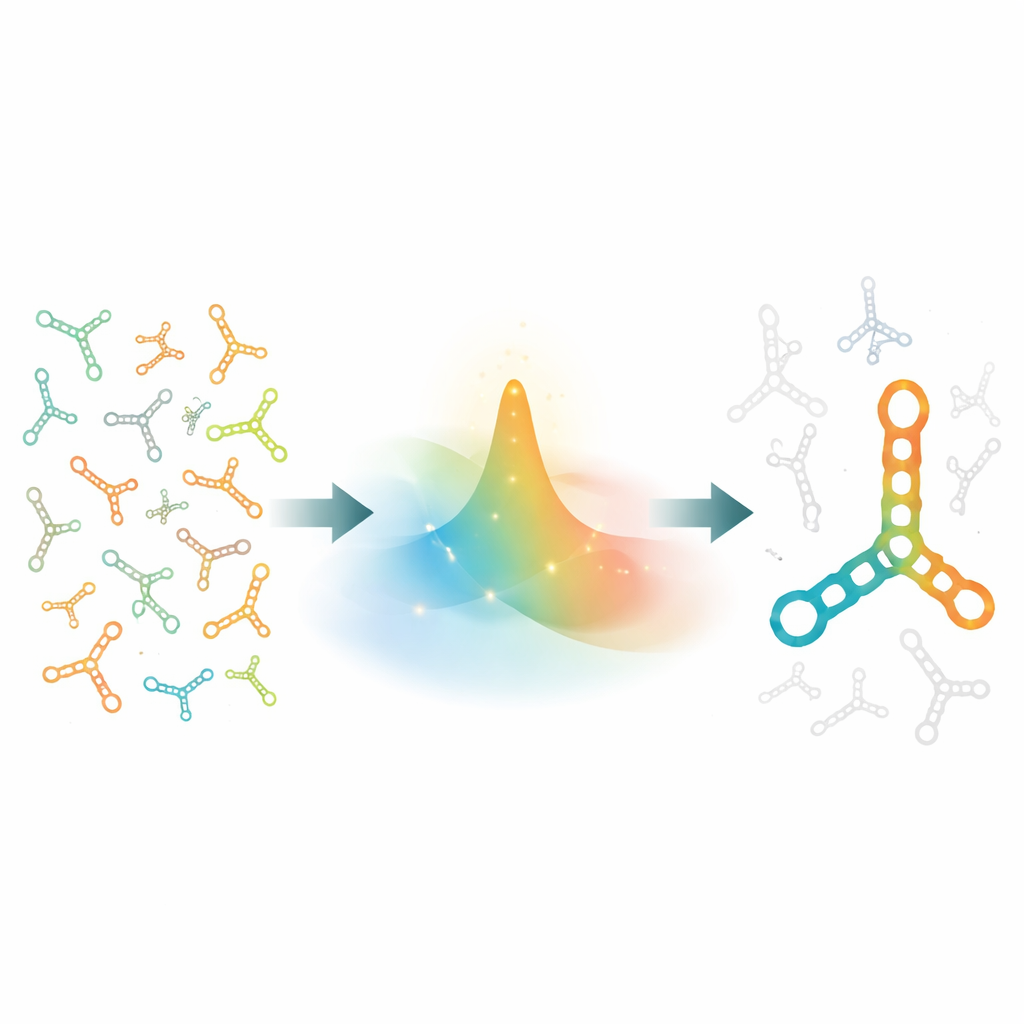
Why designing RNA shapes is so hard
Designing RNA is like trying to pick a string of letters so that, when crumpled up, it forms exactly the right origami figure—and almost nothing else. For a strand of length n, there are 4n possible sequences, so even moderate lengths explode into astronomical possibilities. On top of that, every sequence can fold into an enormous number of alternative shapes that compete with the desired one. A useful design must not only favor the target shape as its lowest-energy form but also make competing shapes much less likely, so that, in a crowd of possibilities, the correct structure dominates. Traditional search methods tweak one or a few positions at a time and quickly bog down in this maze of options, especially for long and complex RNAs.
A new way to explore possibilities at once
Instead of walking from one candidate sequence to the next, SamplingDesign thinks in terms of a whole cloud of possibilities. It starts with a probability distribution spread over all sequences that are compatible with the target shape—that is, sequences whose paired positions can form real chemical base pairs. The method then uses gradient-based optimization, a workhorse of machine learning, to steadily reshape this distribution so that sequences likely to fold well into the target structure gain probability while poor candidates lose it. Crucially, the authors do not optimize a single score for a single sequence; they optimize the average performance of all sequences under the current distribution, which encourages broad exploration early on and fine-tuning later.
Capturing how bases work together
Key to this approach is a more realistic way of representing how positions along the RNA depend on each other. Rather than treating each nucleotide as an independent choice, SamplingDesign bundles certain positions into “coupled variables.” For every base pair, the two partners share a small joint probability table over the six chemically allowed pair types, automatically excluding invalid combinations. Similar coupling is used for neighboring positions that affect each other’s energy, such as mismatch and trimismatch groups around loops. This shrinks the design space to only valid sequences and makes optimization smoother, because updates act directly on meaningful base-pair and mismatch choices rather than on isolated letters.
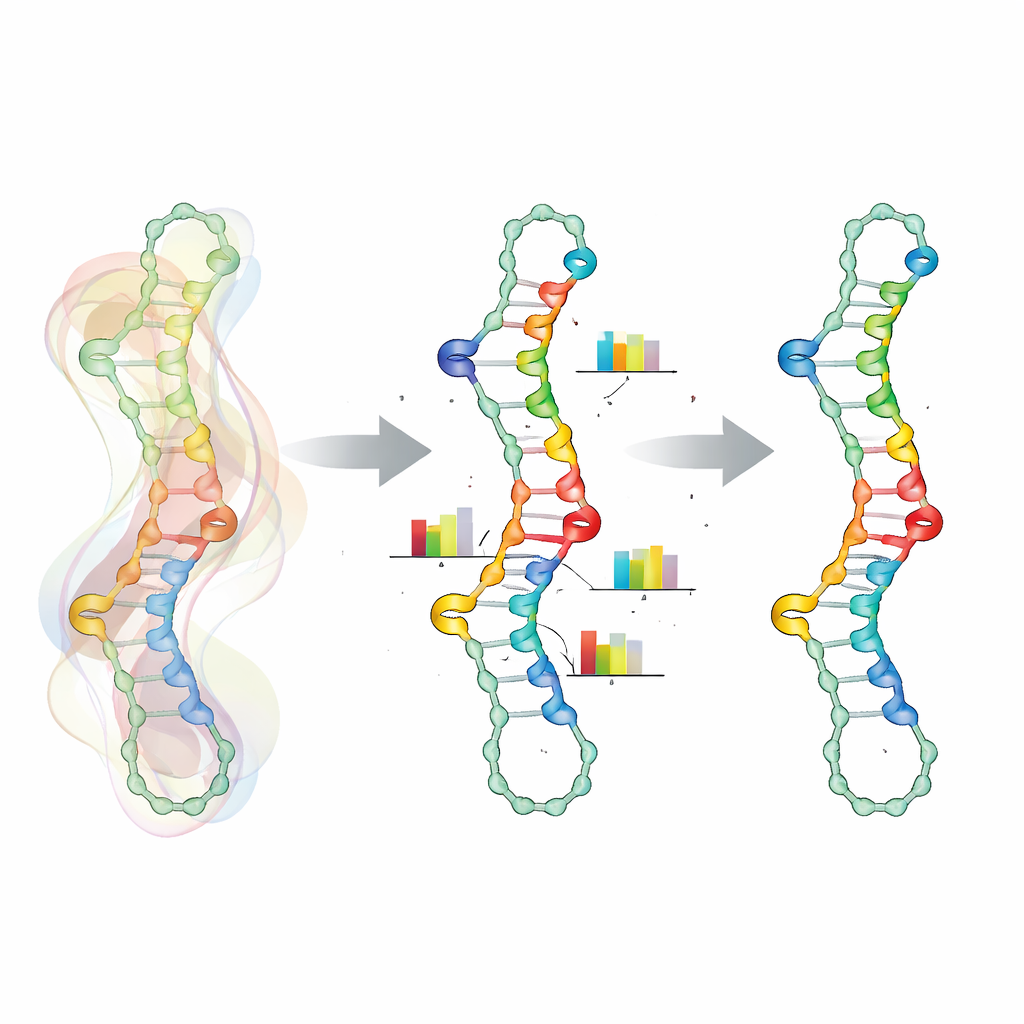
Letting randomness guide smarter choices
Because it is impossible to calculate exact averages over the huge space of sequences and folds, SamplingDesign relies on Monte Carlo sampling. At each step, it draws a manageable number of sequences from the current distribution, evaluates how well each folds according to thermodynamic models, and uses these samples to estimate both the average objective (such as the probability of the target structure) and how to adjust the distribution. Over many iterations, the probability mass shifts toward better sequences and the distribution becomes sharper. Instead of simply taking the single most probable sequence at the end, the method keeps track of all samples it has seen and selects the one that actually performs best by the chosen metric, gaining the benefits of wide exploration without losing focus.
Outperforming existing tools on hard puzzles
The authors tested SamplingDesign on several standard collections of RNA “puzzles,” including the widely used Eterna100 benchmark, which ranges from simple hairpins to long, intricate shapes of up to 400 nucleotides. Across nearly all measures that look at whole ensembles of structures—especially the Boltzmann probability of the target shape and the “ensemble defect,” which tracks how often nucleotides are misfolded—SamplingDesign beat state-of-the-art design tools that rely on local search or simpler continuous methods. The advantage was most striking for the longest and hardest puzzles, where traditional algorithms often get stuck in poor solutions while SamplingDesign continues to find sequences whose target shapes stand out clearly from competitors.
What this means for future RNA technologies
In plain terms, this work shows that treating RNA design as a guided exploration of many sequences at once, rather than a step-by-step editing game, can produce cleaner, more reliable folds—especially for large and challenging targets. By modeling how bases interact in pairs and groups, and by using sampling to navigate an otherwise intractable landscape, SamplingDesign provides a flexible framework that can optimize different design goals. The authors suggest it could be extended to tailor messenger RNAs for vaccines or therapies and to incorporate experimental constraints. As improved computational design meets laboratory testing, such methods could help turn abstract RNA blueprints into practical molecular tools for medicine.
Citation: Tang, W.Y., Dai, N., Zhou, T. et al. SamplingDesign: RNA design via continuous optimization with coupled variables and Monte-Carlo sampling. Nat Commun 17, 2950 (2026). https://doi.org/10.1038/s41467-025-67901-3
Keywords: RNA design, inverse folding, Monte Carlo sampling, continuous optimization, mRNA therapeutics