Clear Sky Science · sv
Hämning av RNA-polymeras II‑aktiverande CDK9 och CDK12/13, men inte av cellcykel‑relevanta CDK:er, inducerar apoptos genom att nedreglera de kortlivade Bcl‑2‑proteinerna Mcl1 och Bfl1/A1
Hur avstängning av cancerns skyddssköld kan döda tumörceller
Cancerceller överlever ofta hårda behandlingar eftersom de slår på kraftfulla interna skyddssköldar som blockerar celldöd. Denna studie ställer en enkel fråga med stora medicinska följder: kan vi stänga av endast de brytare som håller cancerceller vid liv, utan att skada normal celldelning? Forskarna undersökte en familj enzymer kallade CDK:er och fann att endast några få, strikt kopplade till genläsning snarare än cellcykeln, är de verkliga livlinorna för vissa blodcancerceller.
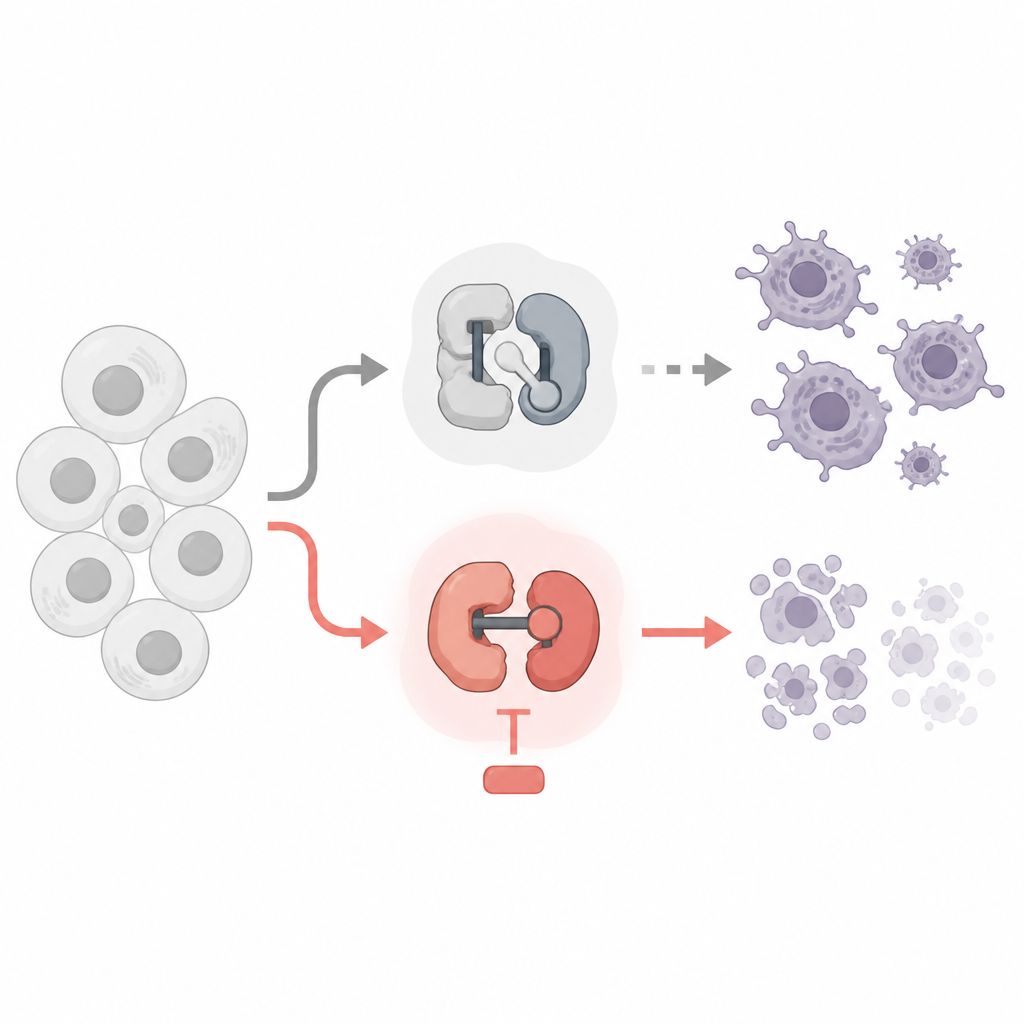
Två slags små brytare inuti våra celler
Varje cell använder cyclin‑beroende kinaser, eller CDK:er, som små molekylära brytare. Vissa CDK:er kontrollerar när en cell växer och delar sig, medan andra styr hur gener läses till RNA och sedan till proteiner. Cancerläkemedel som riktar sig mot CDK:er finns redan, men många utvecklades för att stoppa celldelning. Författarna jämförde läkemedel som blockerar cellcykel‑CDK:er med läkemedel som blockerar genläsnings‑CDK:er, och använde leukemi‑ och lymfom‑cellinjer som testmodell. De frågade vilka brytare som säkert kan stängas av och vilka som verkligen driver cancerceller över gränsen in i programmerad död.
Att stänga ner genläsning, inte celldelning
Teamet upptäckte en slående uppdelning. Läkemedel som stoppade klassiska cellcykel‑CDK:er, såsom CDK1 och CDK4/6, bromsade celldelningen men dödade inte cancercellerna, inte ens efter flera dagar. Överraskande nog utlöste inte heller blockering av CDK7, som hjälper till att starta genläsning, mycket celldöd vid doser som träffade detta mål specifikt. I kontrast ledde mycket selektiva läkemedel mot CDK9 eller CDK12/13, som hjälper enzymet RNA‑polymeras II att läsa igenom gener, till att cancercellerna snabbt genomgick apoptos, en ordnad form av cellulärt självmord. Dessa läkemedel avlägsnade viktiga kemiska märken från RNA‑polymeras II, skar kraftigt ned ny RNA‑produktion och aktiverade cellens interna dödsmaskineri.
Att avväpna cellens livvakter
Varför dödar avstängning av denna fas av genläsningen cellerna? Svaret ligger i en grupp kortlivade ”livvakt”‑proteiner från Bcl‑2‑familjen, särskilt Mcl1 och A1. Dessa proteiner håller normalt dödseffektorernas Bax och Bak i schack vid mitokondrierna, cellens kraftverk. Studien visar att blockering av CDK9 eller CDK12/13 snabbt sänker nivåerna av Mcl1 och A1, medan längre livade skyddsproteiner som Bcl‑2 och Bcl‑xL förblir i stort sett oförändrade. När Mcl1 och A1 försvinner släpps Bax och Bak fria, mitokondrierna läcker pro‑döds‑signaler och apoptos följer. Genetiska experiment bekräftade denna kedja: avlägsnande av Bax och Bak skyddade cellerna, medan extra Bcl‑xL, men inte extra Bcl‑2, kunde blockera död som utlösts av CDK9‑ och CDK12/13‑läkemedlen.
Två samarbetande maskiner och ett kraftfullt läkemedelspar
Även om CDK9 och CDK12/13 båda hjälper RNA‑polymeras II, verkar de på olika hjälpproteiner som kontrollerar skiftet från pausad till fullt aktiv genläsning. Genom att granska proteiner bundna till DNA fann författarna att CDK9 behövs för att släppa ett bromsande komplex och rekrytera faktorer som SPT6, medan CDK12/13 hjälper till att hålla ett annat komplex, PAF1C, stabilt bundet. Att blockera endera stör elongationsfasen på sitt sätt. När forskarna kombinerade en CDK9‑hämmare med en CDK12/13‑hämmare dog cancercellerna mycket effektivare än med något av läkemedlen ensamt, vilket visade stark synergism som enkla transkriptionshämmare inte kunde efterlikna.
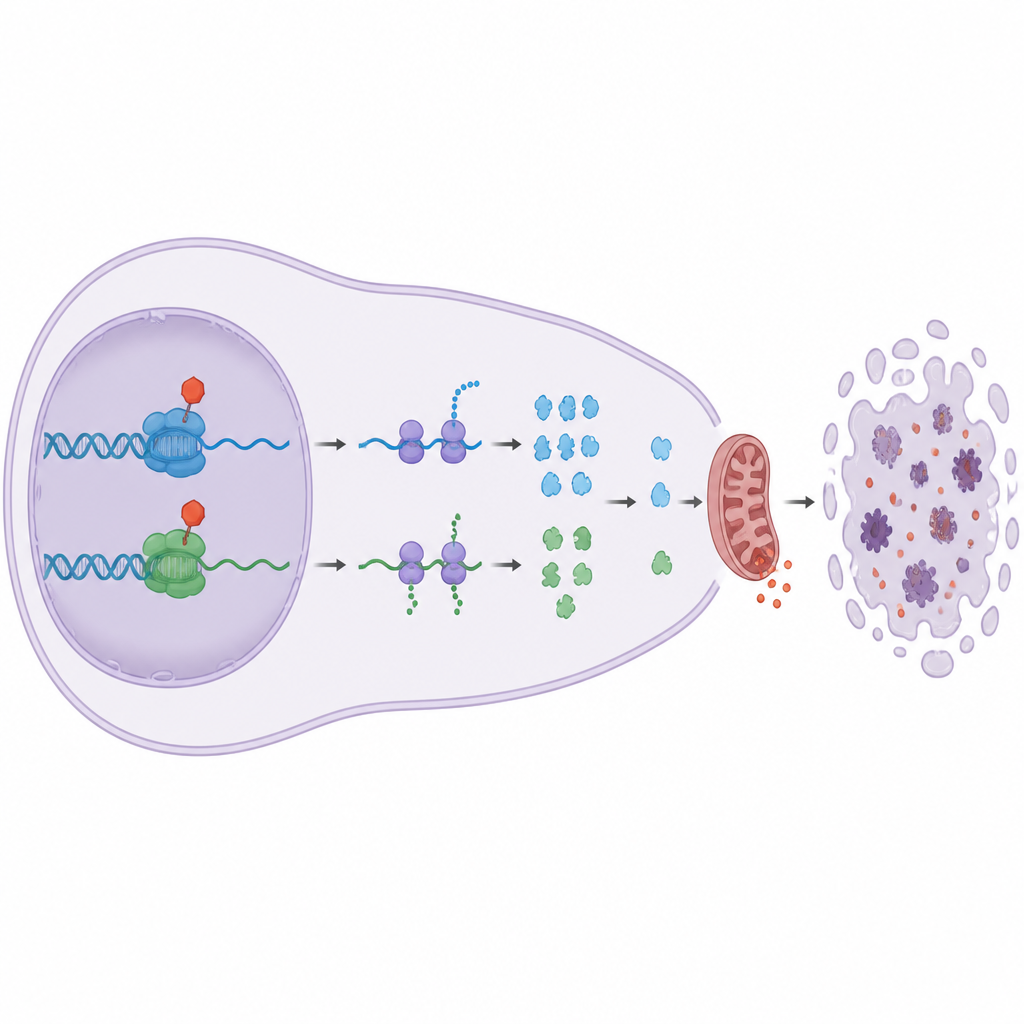
Varför detta är viktigt för framtida cancerbehandling
Många tumörer är beroende av Bcl‑2‑familjens livvakter för att motstå cytostatika, och vissa överproducerar redan Bcl‑2 självt. Denna studie visar att riktade inhibitorer av elongations‑CDK:erna CDK9 och CDK12/13 kan kringgå höga Bcl‑2‑nivåer genom att sänka Mcl1 och A1, som kontrollerar både Bax och Bak. I klartext: dessa läkemedel drar undan flera nyckelsteg i cancercellens säkerhetsstege samtidigt som andra CDK:er som främst styr normal celldelning skonas. Arbetet antyder att finjusterade hämmare av CDK9 och CDK12/13, använda ensamma eller i kombination, kan bilda en kraftfull ny strategi för att driva resistenta blodcancerceller till självdestruktion samtidigt som oönskad skada på frisk vävnad minimeras.
Citering: Krings, K.S., Hatzfeld, J., Weller, S. et al. Inhibition of RNA polymerase II-activating CDK9 and CDK12/13, but not of cell cycle relevant CDKs, induces apoptosis by downregulating the short-lived Bcl-2 proteins Mcl1 and Bfl1/A1. Cell Death Dis 17, 512 (2026). https://doi.org/10.1038/s41419-026-08889-6
Nyckelord: CDK9, CDK12, Mcl1, apoptos, blodcancer