Clear Sky Science · en
Inhibition of RNA polymerase II-activating CDK9 and CDK12/13, but not of cell cycle relevant CDKs, induces apoptosis by downregulating the short-lived Bcl-2 proteins Mcl1 and Bfl1/A1
How turning off cancer’s safety shield can kill tumor cells
Cancer cells often survive harsh treatments because they switch on powerful internal safety shields that block cell death. This study asks a simple question with big medical implications: can we turn off only those switches that keep cancer cells alive, without harming healthy cell division? The researchers explored a family of enzymes called CDKs and found that only a few, tightly linked to gene reading rather than cell division, are the real lifelines for certain blood cancer cells.
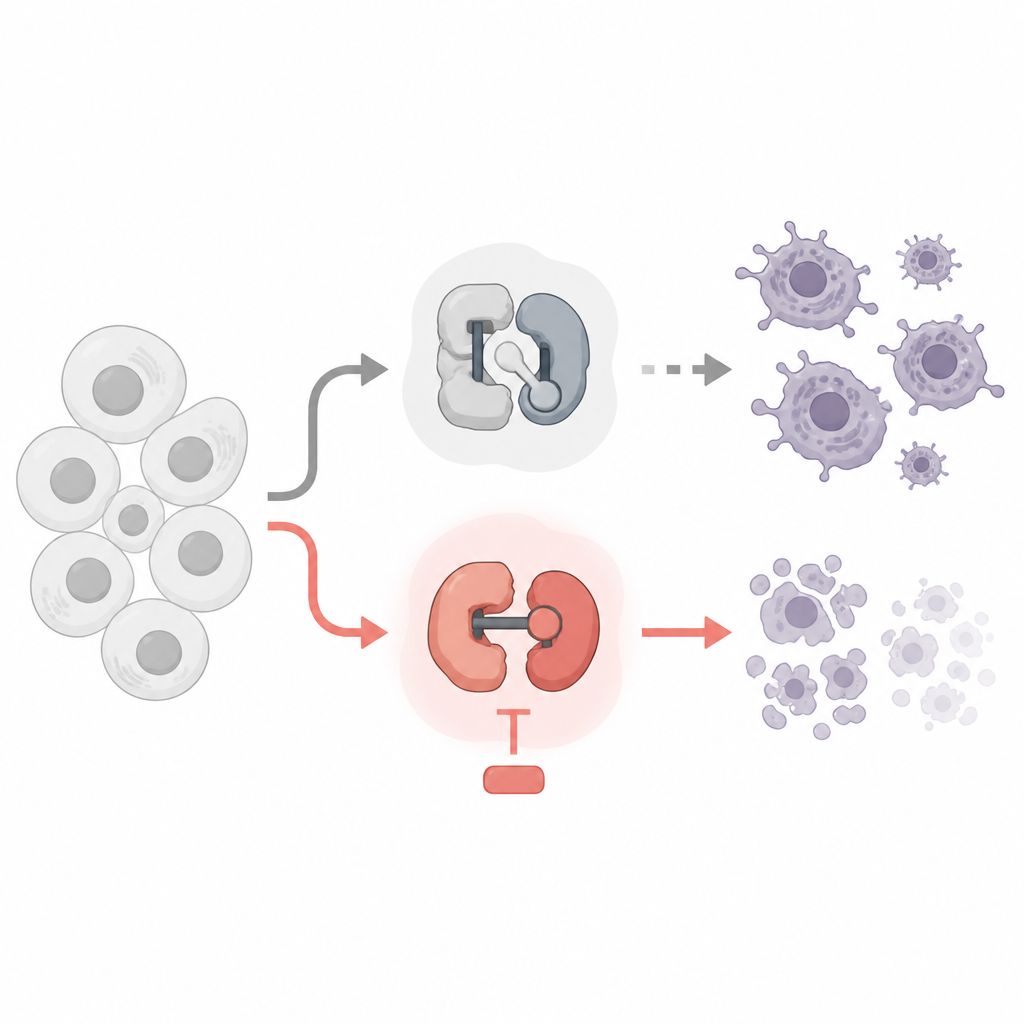
Two kinds of tiny switches inside our cells
Every cell uses cyclin dependent kinases, or CDKs, as tiny molecular switches. Some CDKs control when a cell grows and divides, while others control how genes are read into RNA and then into proteins. Cancer drugs that target CDKs already exist, but many were designed to stop cell division. The authors compared drugs that block cell cycle CDKs with drugs that block gene reading CDKs, using leukemia and lymphoma cell lines as their test bed. They asked which switches can be safely turned off and which ones truly push cancer cells over the edge into programmed death.
Shutting down gene reading, not cell division
The team discovered a striking divide. Drugs that stopped classic cell cycle CDKs, such as CDK1 and CDK4/6, slowed cell division but did not kill the cancer cells, even after several days. Surprisingly, blocking CDK7, which helps start gene reading, also did not trigger much cell death at doses that cleanly hit this target. In contrast, when they used highly selective drugs against CDK9 or CDK12/13, which help an enzyme called RNA polymerase II read genes all the way through, the cancer cells quickly underwent apoptosis, a tidy form of cellular suicide. These drugs stripped key chemical tags from RNA polymerase II, sharply cut new RNA production, and activated the cell’s internal death machinery.
Disarming the cell’s bodyguards
Why does cutting off this phase of gene reading kill the cells? The answer lies in a group of short lived “bodyguard” proteins from the Bcl 2 family, especially Mcl1 and A1. These proteins normally keep the death effectors Bax and Bak in check at the mitochondria, the cell’s power plants. The study shows that blocking CDK9 or CDK12/13 rapidly lowers Mcl1 and A1 levels, while longer lived protectors such as Bcl 2 and Bcl xL remain largely unchanged. When Mcl1 and A1 vanish, Bax and Bak are unleashed, the mitochondria leak pro death signals, and apoptosis follows. Genetic experiments confirmed this chain: removing Bax and Bak protected cells, while extra Bcl xL, but not extra Bcl 2, could block death triggered by the CDK9 and CDK12/13 drugs.
Two cooperating machines and a powerful drug duo
Even though CDK9 and CDK12/13 both help RNA polymerase II, they act on different helper proteins that control the shift from paused to fully active gene reading. By examining proteins attached to DNA, the authors found that CDK9 is needed to release a braking complex and recruit factors like SPT6, while CDK12/13 helps keep another complex, PAF1C, stably attached. Blocking either one disrupts this elongation phase in its own way. When the researchers combined a CDK9 blocker with a CDK12/13 blocker, the cancer cells died far more efficiently than with either drug alone, showing strong synergy that simple transcription blockers could not mimic.
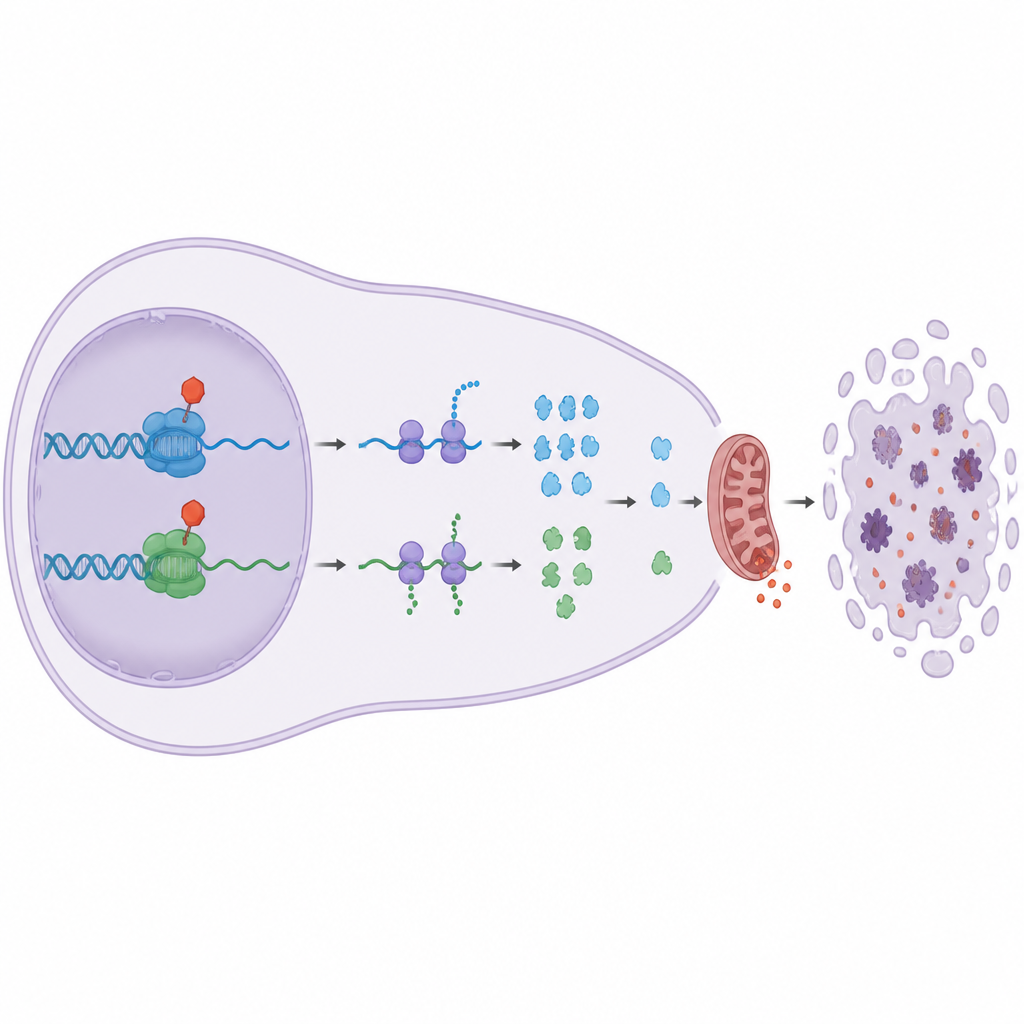
Why this matters for future cancer treatment
Many tumors rely on Bcl 2 family bodyguards to resist chemotherapy, and some already overproduce Bcl 2 itself. This study shows that targeting the elongation CDKs CDK9 and CDK12/13 can bypass high Bcl 2 levels by lowering Mcl1 and A1, which control both Bax and Bak. In plain terms, these drugs pull away several key rungs of the cancer cell’s safety ladder while sparing other CDKs that mainly guide normal cell division. The work suggests that finely tuned inhibitors of CDK9 and CDK12/13, used alone or together, could form a powerful new strategy to push resistant blood cancer cells into self destruction while minimizing unwanted damage to healthy tissue.
Citation: Krings, K.S., Hatzfeld, J., Weller, S. et al. Inhibition of RNA polymerase II-activating CDK9 and CDK12/13, but not of cell cycle relevant CDKs, induces apoptosis by downregulating the short-lived Bcl-2 proteins Mcl1 and Bfl1/A1. Cell Death Dis 17, 512 (2026). https://doi.org/10.1038/s41419-026-08889-6
Keywords: CDK9, CDK12, Mcl1, apoptosis, blood cancer