Clear Sky Science · nl
Remming van RNA-polymerase II-activerende CDK9 en CDK12/13, maar niet van celcyclusrelevante CDK’s, veroorzaakt apoptose door afname van de kortlevende Bcl-2-eiwitten Mcl1 en Bfl1/A1
Hoe het uitschakelen van het veiligheidsschild van kanker tumorcellen kan doden
Kankercellen overleven vaak zware behandelingen omdat ze krachtige interne veiligheidsschilden inschakelen die celdood blokkeren. Deze studie stelt een eenvoudige vraag met grote medische implicaties: kunnen we alleen die schakelaars uitzetten die kankercellen in leven houden, zonder normale celdeling te beschadigen? De onderzoekers onderzochten een familie enzymen die CDK’s worden genoemd en ontdekten dat slechts enkele, nauw verbonden met genlezing in plaats van celdeling, de echte levenslijnen vormen voor bepaalde bloedkankercellen.
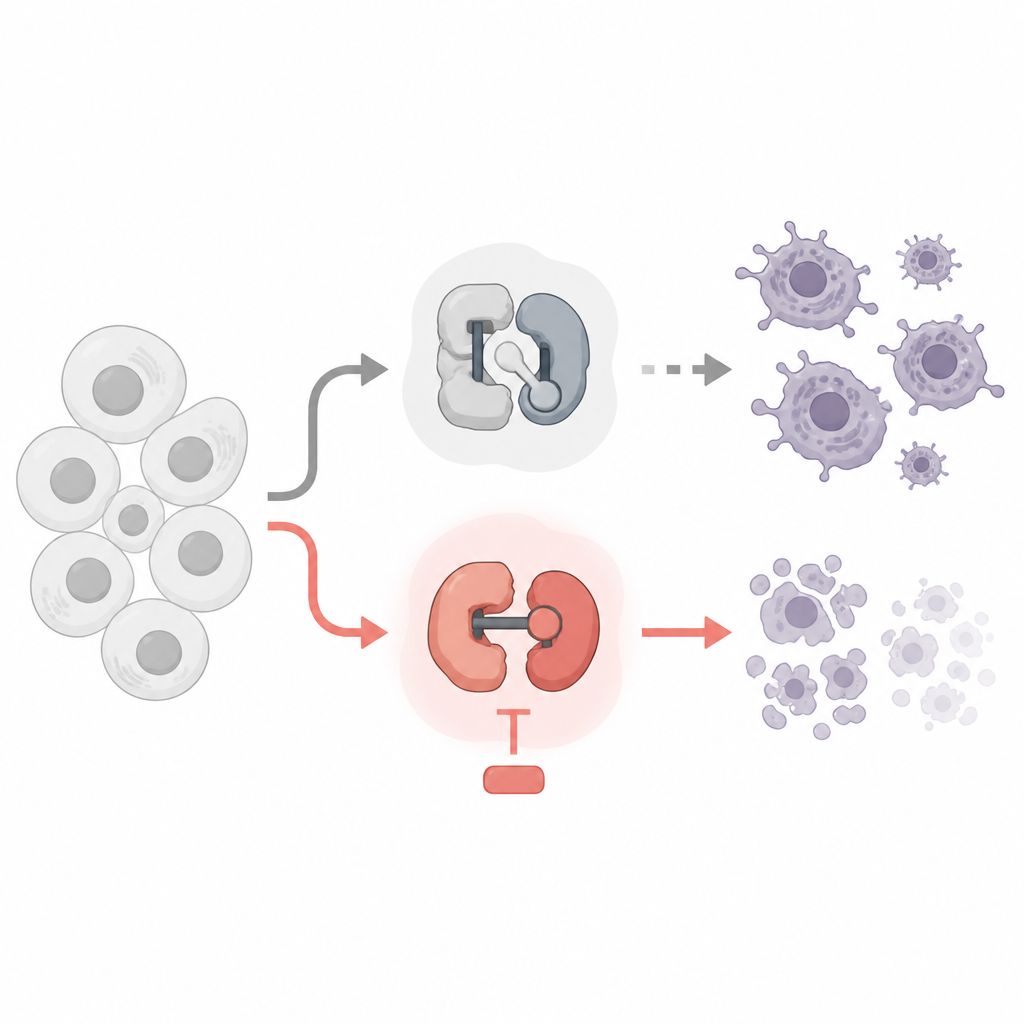
Twee soorten piepkleine schakelaars in onze cellen
Elke cel gebruikt cycline-afhankelijke kinasen, of CDK’s, als piepkleine moleculaire schakelaars. Sommige CDK’s regelen wanneer een cel groeit en deelt, terwijl andere regelen hoe genen worden afgelezen naar RNA en daarna naar eiwitten. Er bestaan al kankermedicijnen die CDK’s remmen, maar veel daarvan zijn ontworpen om celdeling te stoppen. De auteurs vergeleken remmers van celcyclus-CDK’s met remmers van transcriptie-CDK’s, waarbij ze leukemie- en lymfoomcellijnen als proefmodel gebruikten. Ze onderzochten welke schakelaars veilig uit te schakelen zijn en welke schakelaars kankercellen echt over de rand in geprogrammeerde celdood duwen.
Genlezing stilleggen, niet celdeling
Het team ontdekte een opvallende scheidslijn. Middelen die klassieke celcyclus-CDK’s zoals CDK1 en CDK4/6 remden, vertraagden weliswaar de celdeling maar doodden de kankercellen niet, zelfs niet na meerdere dagen. Verrassend genoeg veroorzaakte ook remming van CDK7, dat helpt bij het starten van genlezing, weinig celdood bij concentraties die dit doel specifiek raken. Daarentegen veroorzaakten zeer selectieve remmers van CDK9 of CDK12/13, die een enzym genaamd RNA-polymerase II helpen om genen volledig af te lezen, snel apoptose in de kankercellen. Deze middelen verwijderden sleutelchemische labels van RNA-polymerase II, verminderden scherp de nieuwe RNA-productie en activeerden het interne dode-machinerie van de cel.
De lijfwachten van de cel ontwapenen
Waarom doodt het afsluiten van deze fase van genlezing de cellen? Het antwoord ligt bij een groep kortlevende “lijfwacht”-eiwitten uit de Bcl-2-familie, met name Mcl1 en A1. Deze eiwitten houden normaal de death-effectoren Bax en Bak onder controle bij de mitochondriën, de energiecentrales van de cel. De studie toont aan dat blokkade van CDK9 of CDK12/13 snel leidt tot verlaging van Mcl1- en A1-niveaus, terwijl langerlevende beschermers zoals Bcl-2 en Bcl-xL grotendeels onveranderd blijven. Wanneer Mcl1 en A1 verdwijnen, worden Bax en Bak losgelaten, lekken de mitochondriën pro-doodsignalen en volgt apoptose. Genetische experimenten bevestigden deze keten: het verwijderen van Bax en Bak beschermde cellen, terwijl extra Bcl-xL, maar niet extra Bcl-2, de door CDK9- en CDK12/13-remmers geïnduceerde celdood kon blokkeren.
Twee samenwerkende machines en een krachtig medicijnduo
Hoewel CDK9 en CDK12/13 allebei RNA-polymerase II ondersteunen, werken ze via verschillende hulpproteïnen die de overgang van gepauzeerde naar volledig actieve genlezing regelen. Door eiwitten die aan DNA gebonden zijn te onderzoeken, vonden de auteurs dat CDK9 nodig is om een remmend complex los te laten en factoren zoals SPT6 te rekruteren, terwijl CDK12/13 helpt om een ander complex, PAF1C, stabiel vast te houden. Het blokkeren van elk van beide verstoort deze elongatiefase op zijn eigen manier. Wanneer de onderzoekers een CDK9-remmer combineerden met een CDK12/13-remmer, stierven de kankercellen veel efficiënter dan bij elk middel afzonderlijk, wat sterke synergie liet zien die eenvoudige transcriptieremmers niet konden nabootsen.
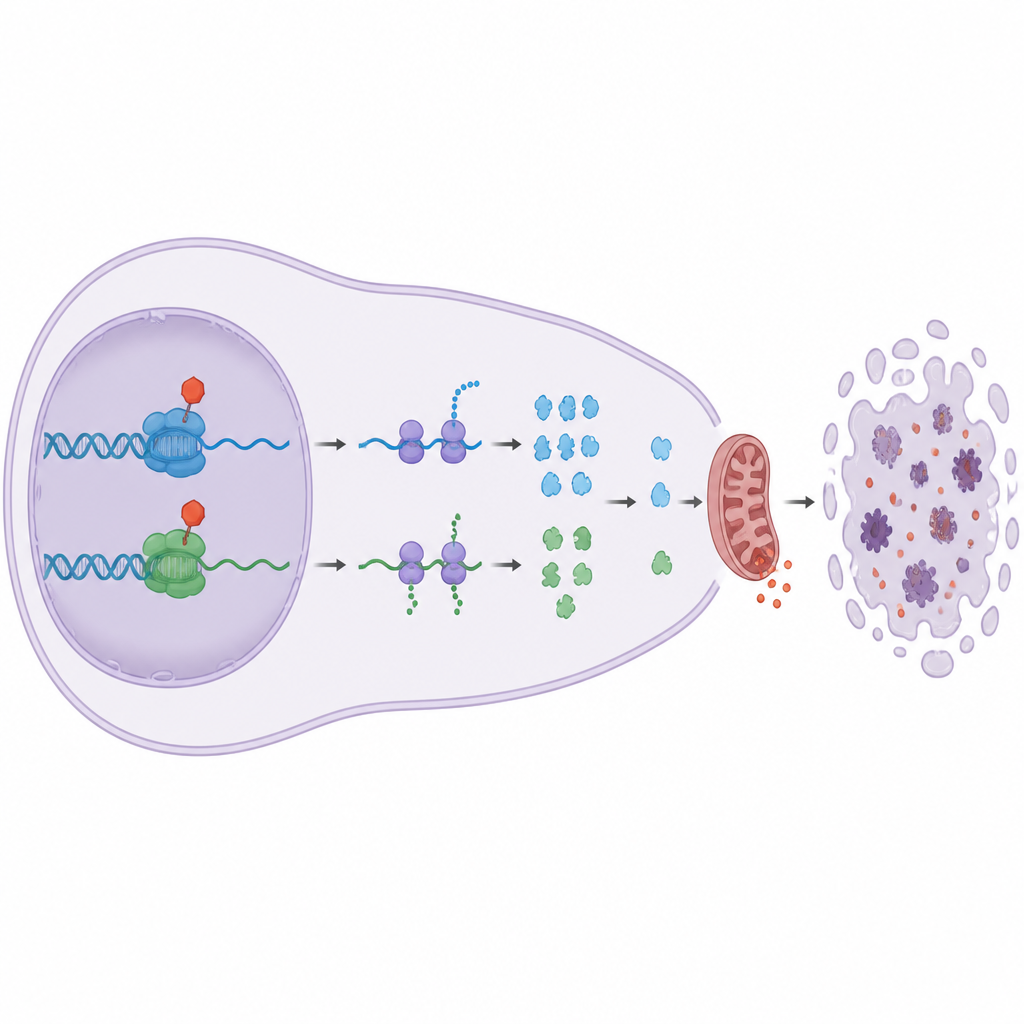
Waarom dit belangrijk is voor toekomstige kankerbehandeling
Veel tumoren vertrouwen op Bcl-2-familie-lijfwachten om chemotherapie te weerstaan, en sommige produceren al te veel Bcl-2 zelf. Deze studie toont dat het richten op de elongatie-CDK’s CDK9 en CDK12/13 hoge Bcl-2-niveaus kan omzeilen door Mcl1 en A1 te verlagen, die zowel Bax als Bak controleren. Simpel gezegd trekken deze middelen meerdere belangrijke sporten van de veiligheidstrap van de kankercel weg, terwijl ze andere CDK’s die voornamelijk normale celdeling sturen sparen. Het werk suggereert dat fijn afgestelde remmers van CDK9 en CDK12/13, afzonderlijk of gecombineerd, een krachtig nieuwe strategie kunnen vormen om resistente bloedkankercellen tot zelfvernietiging te dwingen met minimale schade aan gezond weefsel.
Bronvermelding: Krings, K.S., Hatzfeld, J., Weller, S. et al. Inhibition of RNA polymerase II-activating CDK9 and CDK12/13, but not of cell cycle relevant CDKs, induces apoptosis by downregulating the short-lived Bcl-2 proteins Mcl1 and Bfl1/A1. Cell Death Dis 17, 512 (2026). https://doi.org/10.1038/s41419-026-08889-6
Trefwoorden: CDK9, CDK12, Mcl1, apoptose, bloedkanker