Clear Sky Science · pl
Inhibicja aktywujących polimerazę RNA II CDK9 i CDK12/13, a nie cyklozależnych kinaz związanych z cyklem komórkowym, wywołuje apoptozę przez obniżenie poziomu krótkotrwałych białek Bcl-2 Mcl1 i Bfl1/A1
Jak wyłączenie tarczy ochronnej raka może zabić komórki guzowe
Komórki nowotworowe często przetrwają ostre leczenie, ponieważ włączają silne wewnętrzne tarcze ochronne, które blokują śmierć komórki. W badaniu postawiono proste pytanie o dalekosiężnych implikacjach medycznych: czy można wyłączyć wyłącznie te przełączniki, które utrzymują przy życiu komórki nowotworowe, bez uszkadzania prawidłowego podziału komórek? Naukowcy badali rodzinę enzymów zwanych CDK i odkryli, że tylko niektóre z nich, ściśle związane z odczytem genów, a nie z podziałem komórkowym, są rzeczywistymi lifeline’ami dla pewnych komórek nowotworów krwi.
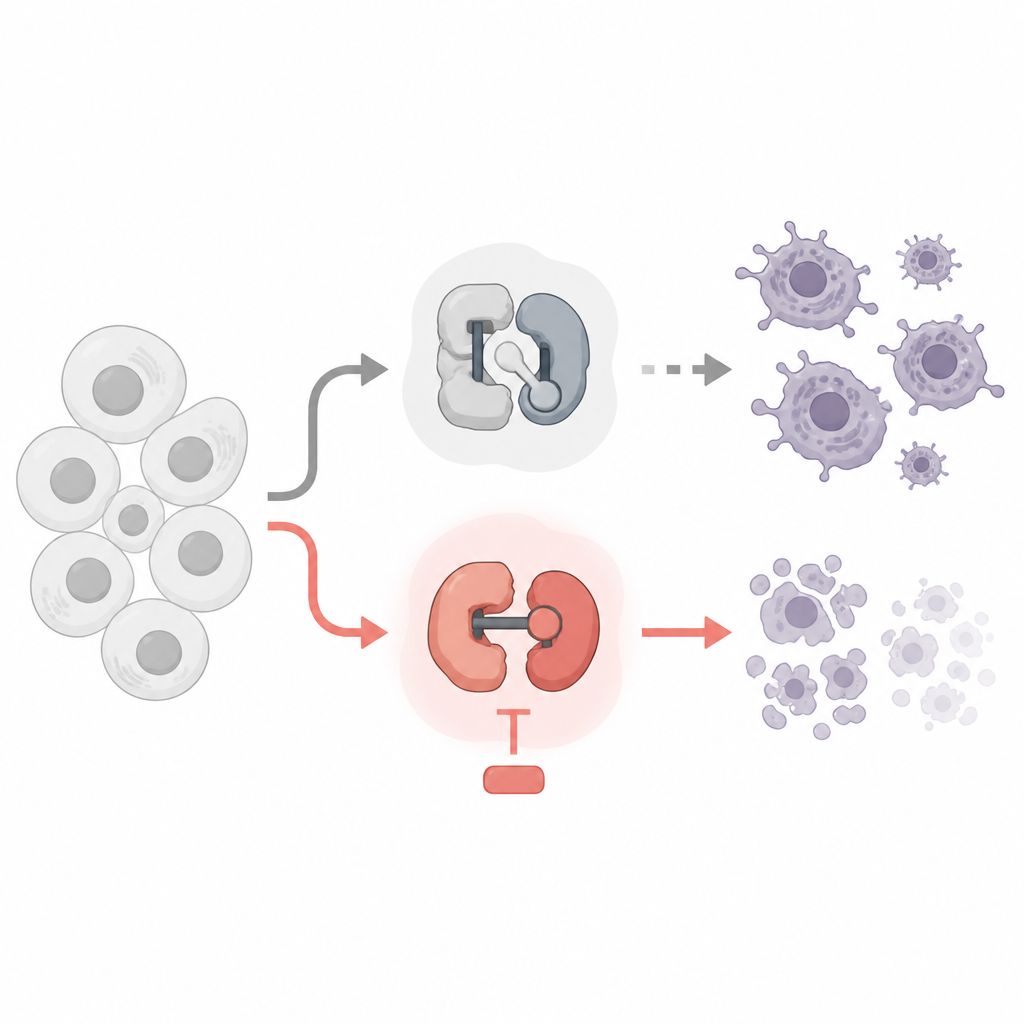
Dwa rodzaje maleńkich przełączników wewnątrz naszych komórek
Każda komórka wykorzystuje cyklinozależne kinazy, czyli CDK, jako maleńkie molekularne przełączniki. Niektóre CDK kontrolują, kiedy komórka rośnie i się dzieli, inne zaś kontrolują, jak geny są odczytywane na RNA, a następnie tłumaczone na białka. Leki przeciwnowotworowe celujące w CDK już istnieją, ale wiele z nich zaprojektowano, by zatrzymywać podział komórek. Autorzy porównali leki blokujące cykliczne CDK biorące udział w cyklu komórkowym z lekami hamującymi transkrypcyjne CDK, używając linii komórkowych białaczkowych i chłoniakowych jako modelu. Badali, które przełączniki można bezpiecznie wyłączyć, a które rzeczywiście popychają komórki nowotworowe w stronę zaprogramowanej śmierci.
Wyłączenie odczytu genów, a nie podziału komórek
Zespół odkrył wyraźne rozróżnienie. Leki zatrzymujące klasyczne CDK cyklu komórkowego, takie jak CDK1 i CDK4/6, spowalniały podziały komórek, ale nie zabijały komórek nowotworowych, nawet po kilku dniach. Co zaskakujące, hamowanie CDK7, który pomaga w inicjacji transkrypcji, również nie wywoływało istotnej śmierci komórek przy dawkach selektywnie uderzających w ten cel. W przeciwieństwie do tego, gdy zastosowano wysoko selektywne inhibitory CDK9 lub CDK12/13, które pomagają enzymowi RNA polimerazie II w pełnym odczycie genów, komórki nowotworowe szybko przeszły w apoptozę — uporządkowaną formę samobójczej śmierci komórki. Leki te usunęły kluczowe modyfikacje z RNA polimerazy II, ostro ograniczyły syntezę nowego RNA i aktywowały wewnętrzne mechanizmy śmierci komórkowej.
Rozbrajanie ochroniarzy komórki
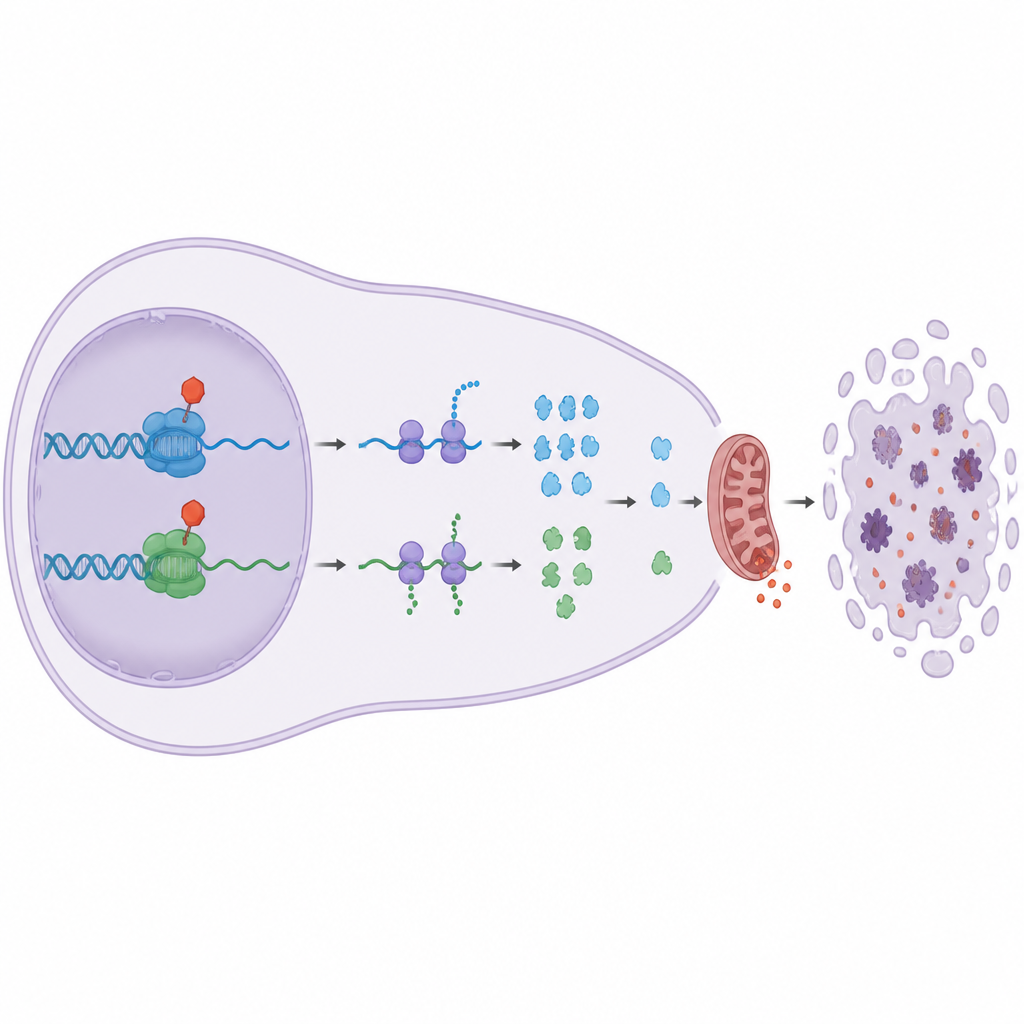
Dlaczego odcięcie tego etapu odczytu genów zabija komórki? Odpowiedź tkwi w grupie krótkotrwałych „ochroniarzy” z rodziny Bcl-2, szczególnie Mcl1 i A1. Białka te normalnie powstrzymują efektory śmierci Bax i Bak przy mitochondriach, elektrowniach komórki. Badanie pokazuje, że blokada CDK9 lub CDK12/13 szybko obniża poziomy Mcl1 i A1, podczas gdy dłużej utrzymujące się protektory, takie jak Bcl-2 i Bcl-xL, pozostają w dużej mierze niezmienione. Kiedy Mcl1 i A1 znikają, Bax i Bak zostają uwolnione, mitochondria zaczynają przeciekać sygnały prośmierci i następuje apoptoza. Eksperymenty genetyczne potwierdziły ten łańcuch zdarzeń: usunięcie Bax i Bak chroniło komórki, a dodatkowa ekspresja Bcl-xL, lecz nie Bcl-2, mogła zablokować śmierć wywołaną przez leki na CDK9 i CDK12/13.
Dwie współpracujące maszyny i silne duo leków
Chociaż CDK9 i CDK12/13 oba wspomagają RNA polimerazę II, działają na różne białka pomocnicze, które kontrolują przejście od zatrzymanego do w pełni aktywnego odczytu genów. Analiza białek związanych z DNA wykazała, że CDK9 jest potrzebny do uwolnienia kompleksu hamującego i rekrutacji czynników takich jak SPT6, podczas gdy CDK12/13 pomaga utrzymać stabilne przyłączenie innego kompleksu, PAF1C. Zablokowanie któregokolwiek z nich zaburza fazę elongacji na swój sposób. Gdy badacze połączyli inhibitor CDK9 z inhibitorem CDK12/13, komórki nowotworowe ginęły znacznie wydajniej niż przy którymkolwiek z leków osobno, co świadczy o silnej synergii, której nie dały się odtworzyć proste inhibitory transkrypcji.
Dlaczego to ma znaczenie dla przyszłego leczenia raka
Wiele guzów polega na ochroniarzach z rodziny Bcl-2, by opierać się chemioterapii, a niektóre już nadprodukują samo Bcl-2. To badanie pokazuje, że ukierunkowanie na elongacyjne CDK — CDK9 i CDK12/13 — może obejść wysoki poziom Bcl-2 przez obniżenie Mcl1 i A1, które kontrolują zarówno Bax, jak i Bak. Mówiąc prosto, leki te zabierają kilka kluczowych szczebli z drabiny bezpieczeństwa komórki nowotworowej, jednocześnie oszczędzając inne CDK, które głównie kierują normalnym podziałem komórek. Praca sugeruje, że precyzyjnie dobrane inhibitory CDK9 i CDK12/13, stosowane osobno lub łącznie, mogą stworzyć potężną nową strategię popychającą oporne komórki białaczkowe do samounicestwienia przy minimalizowaniu niepożądanego uszkodzenia zdrowej tkanki.
Cytowanie: Krings, K.S., Hatzfeld, J., Weller, S. et al. Inhibition of RNA polymerase II-activating CDK9 and CDK12/13, but not of cell cycle relevant CDKs, induces apoptosis by downregulating the short-lived Bcl-2 proteins Mcl1 and Bfl1/A1. Cell Death Dis 17, 512 (2026). https://doi.org/10.1038/s41419-026-08889-6
Słowa kluczowe: CDK9, CDK12, Mcl1, apoptoza, nowotwór krwi