Clear Sky Science · es
Inhibición de las CDK9 y CDK12/13 que activan la ARN polimerasa II, pero no de las CDK relevantes para el ciclo celular, induce apoptosis al reducir las proteínas Bcl-2 de vida corta Mcl1 y Bfl1/A1
Cómo apagar el escudo de protección del cáncer puede matar a las células tumorales
Las células cancerosas a menudo sobreviven a tratamientos agresivos porque activan potentes escudos internos que bloquean la muerte celular. Este estudio plantea una pregunta sencilla con grandes implicaciones médicas: ¿podemos apagar únicamente esos interruptores que mantienen vivas a las células cancerosas, sin dañar la división celular sana? Los investigadores exploraron una familia de enzimas llamadas CDK y hallaron que solo unas pocas, estrechamente vinculadas a la lectura de genes más que a la división celular, son las verdaderas vías de supervivencia para ciertos cánceres de la sangre.
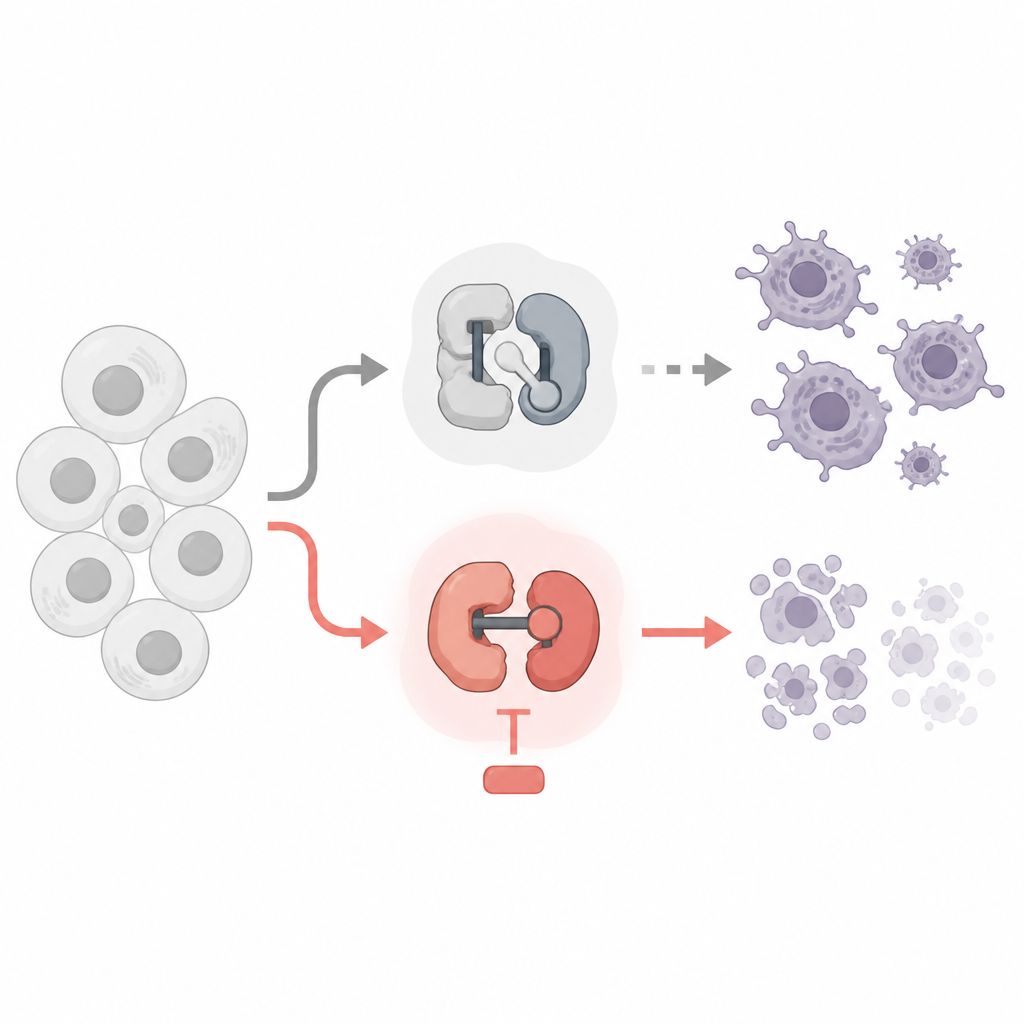
Dos tipos de diminutos interruptores dentro de nuestras células
Cada célula utiliza quininas dependientes de ciclina, o CDK, como pequeños interruptores moleculares. Algunas CDK controlan cuándo una célula crece y se divide, mientras que otras controlan cómo se leen los genes en ARN y luego en proteínas. Ya existen fármacos oncológicos que apuntan a CDK, pero muchos se diseñaron para detener la división celular. Los autores compararon fármacos que bloquean las CDK del ciclo celular con fármacos que bloquean las CDK de la lectura génica, usando líneas celulares de leucemia y linfoma como banco de pruebas. Preguntaron qué interruptores se pueden apagar con seguridad y cuáles realmente empujan a las células cancerosas al borde de la muerte programada.
Apagar la lectura génica, no la división celular
El equipo descubrió una división notable. Los fármacos que detuvieron las CDK clásicas del ciclo celular, como CDK1 y CDK4/6, ralentizaron la división celular pero no mataron a las células cancerosas, ni siquiera tras varios días. Sorprendentemente, bloquear CDK7, que ayuda a iniciar la lectura génica, tampoco desencadenó mucha muerte celular a dosis que impactaban limpiamente este objetivo. En contraste, cuando usaron fármacos altamente selectivos contra CDK9 o CDK12/13, que ayudan a una enzima llamada ARN polimerasa II a leer los genes hasta el final, las células cancerosas rápidamente entraron en apoptosis, una forma ordenada de suicidio celular. Estos fármacos eliminaron marcas químicas clave de la ARN polimerasa II, redujeron drásticamente la producción de ARN nuevo y activaron la maquinaria interna de muerte celular.
Desarmando a los guardianes de la célula
¿Por qué cortar esta fase de la lectura génica mata a las células? La respuesta reside en un grupo de proteínas “guardianas” de vida corta de la familia Bcl-2, en especial Mcl1 y A1. Estas proteínas habitualmente mantienen a raya a los ejecutores de la muerte Bax y Bak en las mitocondrias, las centrales energéticas de la célula. El estudio muestra que bloquear CDK9 o CDK12/13 reduce rápidamente los niveles de Mcl1 y A1, mientras que protectores de vida más larga como Bcl-2 y Bcl-xL permanecen en gran medida sin cambios. Cuando Mcl1 y A1 desaparecen, Bax y Bak se liberan, las mitocondrias filtran señales pro-muerte y sigue la apoptosis. Experimentos genéticos confirmaron esta cadena: eliminar Bax y Bak protegió a las células, mientras que un exceso de Bcl-xL, pero no un exceso de Bcl-2, pudo bloquear la muerte desencadenada por los fármacos contra CDK9 y CDK12/13.
Dos máquinas que cooperan y un potente dúo de fármacos
Aunque CDK9 y CDK12/13 ayudan ambas a la ARN polimerasa II, actúan sobre factores auxiliares distintos que controlan el cambio de una lectura génica en pausa a una lectura completamente activa. Al examinar proteínas unidas al ADN, los autores hallaron que CDK9 es necesario para liberar un complejo de frenado y reclutar factores como SPT6, mientras que CDK12/13 ayuda a mantener otro complejo, PAF1C, firmemente unido. Bloquear cualquiera de los dos interrumpe esta fase de elongación a su manera. Cuando los investigadores combinaron un inhibidor de CDK9 con uno de CDK12/13, las células cancerosas murieron con mucha más eficiencia que con cualquiera de los fármacos por separado, mostrando una fuerte sinergia que los simples bloqueadores de transcripción no pudieron imitar.
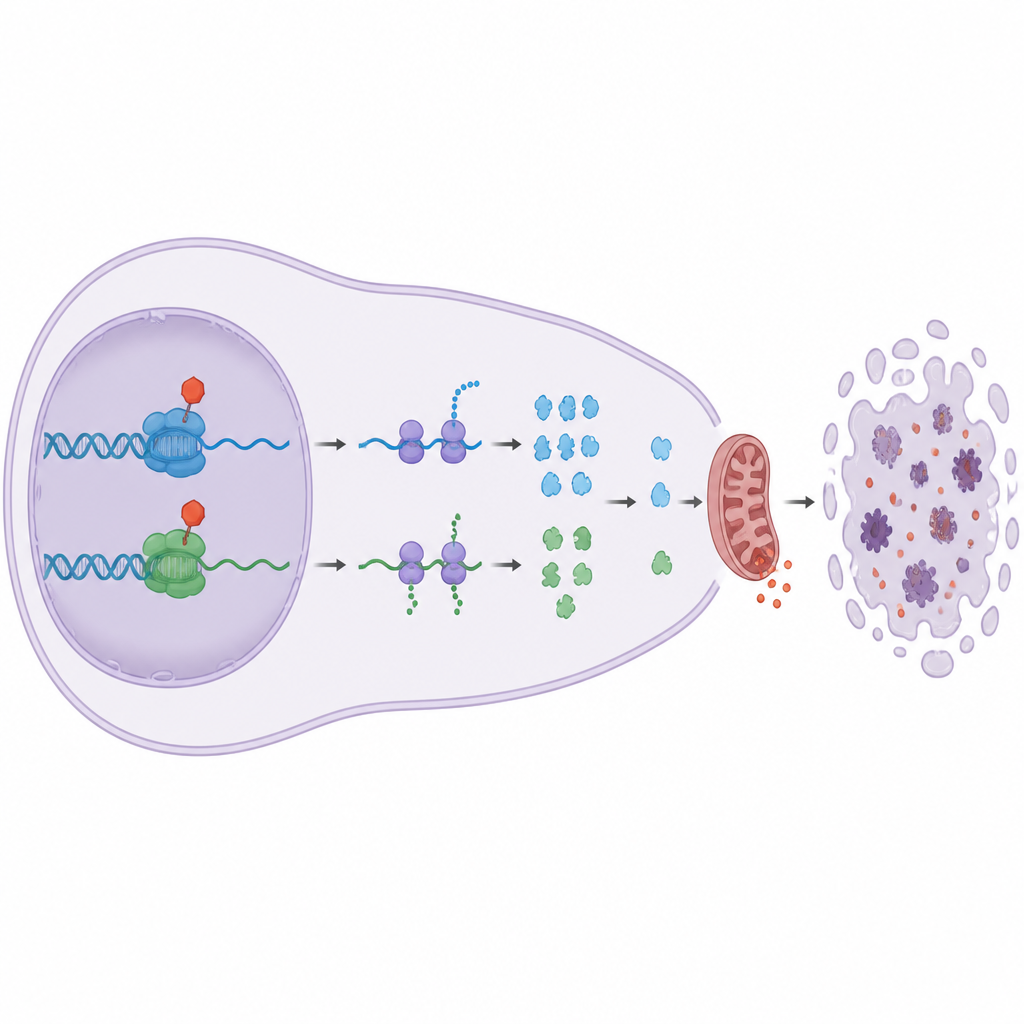
Por qué esto importa para el tratamiento futuro del cáncer
Muchos tumores dependen de las proteínas guardianas de la familia Bcl-2 para resistir la quimioterapia, y algunos ya sobreproducen Bcl-2. Este estudio muestra que dirigir la diana a las CDK de elongación CDK9 y CDK12/13 puede sortear niveles altos de Bcl-2 al reducir Mcl1 y A1, que controlan tanto a Bax como a Bak. En términos sencillos, estos fármacos quitan varios peldaños clave de la escalera de seguridad de la célula cancerosa mientras respetan otras CDK que guían principalmente la división celular normal. El trabajo sugiere que inhibidores finamente ajustados de CDK9 y CDK12/13, usados solos o en combinación, podrían constituir una nueva estrategia potente para empujar a las células de cáncer sanguíneo resistentes hacia la autodestrucción minimizando el daño no deseado al tejido sano.
Cita: Krings, K.S., Hatzfeld, J., Weller, S. et al. Inhibition of RNA polymerase II-activating CDK9 and CDK12/13, but not of cell cycle relevant CDKs, induces apoptosis by downregulating the short-lived Bcl-2 proteins Mcl1 and Bfl1/A1. Cell Death Dis 17, 512 (2026). https://doi.org/10.1038/s41419-026-08889-6
Palabras clave: CDK9, CDK12, Mcl1, apoptosis, cáncer hematológico