Clear Sky Science · de
Hemmung der RNA-Polymerase-II-aktivierenden CDK9 und CDK12/13, nicht aber zellzyklusrelevanter CDKs, löst Apoptose durch Herunterregulieren der kurzlebigen Bcl-2-Proteine Mcl1 und Bfl1/A1 aus
Wie das Abschalten des Schutzschilds von Krebs Tumorzellen töten kann
Krebszellen überleben oft harte Behandlungen, weil sie leistungsstarke interne Schutzschilde aktivieren, die den Zelltod blockieren. Diese Studie stellt eine einfache Frage mit großen medizinischen Folgen: Kann man nur jene Schalter ausschalten, die Krebszellen am Leben erhalten, ohne die normale Zellteilung zu schädigen? Die Forscher untersuchten eine Enzymfamilie namens CDKs und fanden, dass nur wenige, die eng mit dem Ablesen von Genen und nicht mit der Zellteilung verknüpft sind, die eigentlichen Lebensadern bestimmter Blutkrebszellen darstellen.
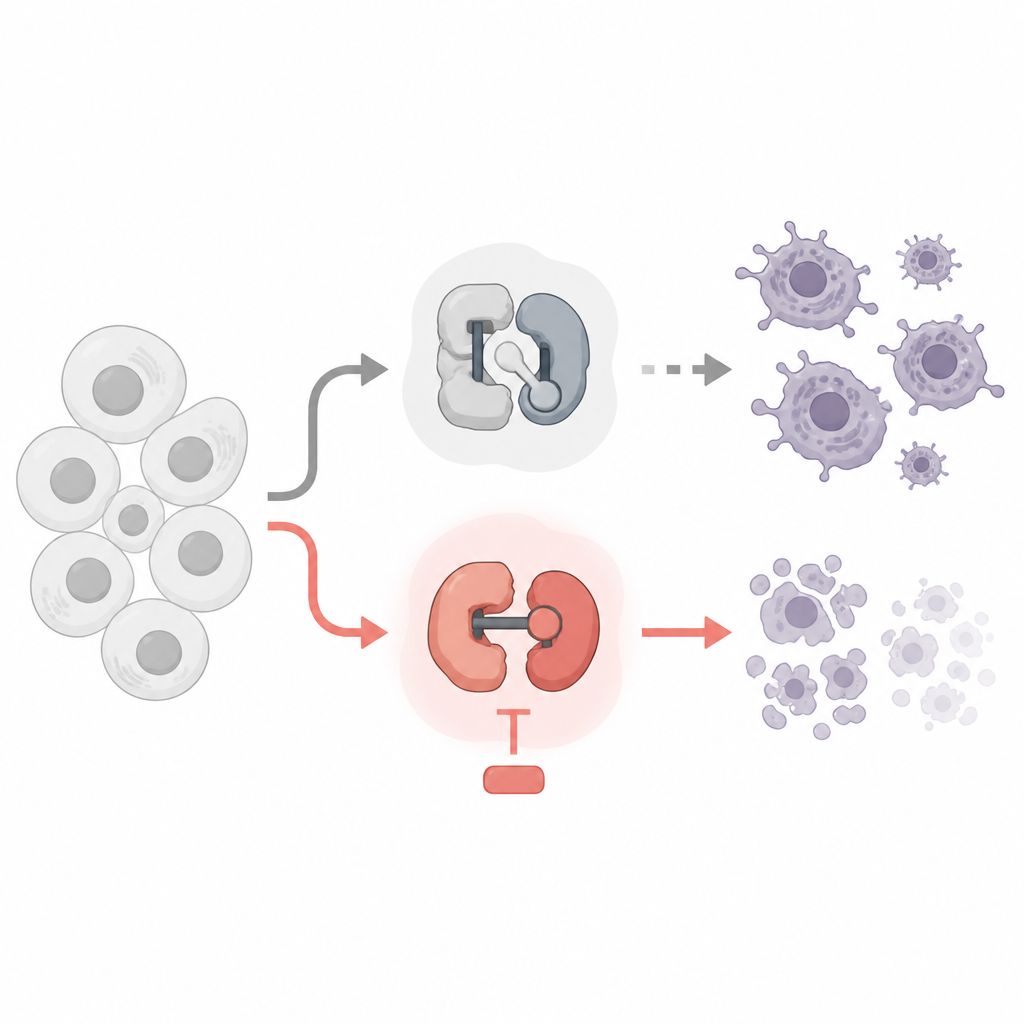
Zwei Arten winziger Schalter in unseren Zellen
Jede Zelle nutzt cyclinabhängige Kinasen, oder CDKs, als winzige molekulare Schalter. Manche CDKs steuern, wann eine Zelle wächst und sich teilt, andere bestimmen, wie Gene in RNA und dann in Proteine umgesetzt werden. Es gibt bereits Krebsmedikamente, die CDKs angreifen, viele wurden jedoch entwickelt, um die Zellteilung zu stoppen. Die Autoren verglichen Wirkstoffe, die Zellzyklus-CDKs hemmen, mit solchen, die transkriptionelle CDKs blockieren, und verwendeten Leukämie- und Lymphomzelllinien als Versuchssystem. Sie wollten wissen, welche Schalter gefahrlos abgeschaltet werden können und welche tatsächlich Krebszellen zur programmierten Selbsttötung treiben.
Das Abstellen des Gablesens, nicht der Zellteilung
Das Team entdeckte eine markante Trennung. Wirkstoffe, die klassische Zellzyklus-CDKs wie CDK1 und CDK4/6 blockierten, verlangsamten zwar die Zellteilung, töteten die Krebszellen aber nicht, selbst nach mehreren Tagen. Überraschenderweise löste auch die Hemmung von CDK7, das beim Start des Gablesens hilft, bei zielgerichteten Dosen nur wenig Zelltod aus. Im Gegensatz dazu führten hochselektive Hemmstoffe gegen CDK9 oder CDK12/13, die der RNA-Polymerase II helfen, Gene vollständig abzulesen, schnell zur Apoptose der Krebszellen — eine ordentliche Form des zellulären Selbstmords. Diese Wirkstoffe entfernten bestimmte chemische Markierungen von RNA-Polymerase II, reduzierten die Neubildung von RNA drastisch und aktivierten die innere Todesmaschinerie der Zelle.
Entwaffnung der Körperwächter der Zelle
Warum tötet das Abschneiden dieser Phase des Gablesens die Zellen? Die Antwort liegt in einer Gruppe kurzlebiger „Körperwächter“-Proteine der Bcl-2-Familie, insbesondere Mcl1 und A1. Diese Proteine halten normalerweise die Todesauslöser Bax und Bak an den Mitochondrien, den Kraftwerken der Zelle, in Schach. Die Studie zeigt, dass die Hemmung von CDK9 oder CDK12/13 die Mcl1- und A1-Spiegel rasch senkt, während länger lebende Schutzproteine wie Bcl-2 und Bcl-xL weitgehend unverändert bleiben. Wenn Mcl1 und A1 verschwinden, werden Bax und Bak aktiviert, die Mitochondrien setzen pro-Tod-Signale frei und es folgt die Apoptose. Genetische Experimente bestätigten diese Kette: Das Entfernen von Bax und Bak schützte die Zellen, während zusätzliches Bcl-xL — nicht aber zusätzliches Bcl-2 — den durch CDK9- und CDK12/13-Inhibitoren ausgelösten Zelltod blockieren konnte.
Zwei kooperierende Maschinen und ein kräftiges Wirkstoffduo
Obwohl CDK9 und CDK12/13 beide der RNA-Polymerase II helfen, wirken sie über unterschiedliche Hilfsproteine, die den Übergang vom pausierten zum voll aktiven Gablesen steuern. Durch Untersuchung von Proteinen, die an die DNA gebunden sind, fanden die Autoren heraus, dass CDK9 nötig ist, um einen Bremskomplex zu lösen und Faktoren wie SPT6 anzuwerben, während CDK12/13 hilft, einen anderen Komplex, PAF1C, stabil zu halten. Die Blockade des einen oder anderen stört diese Elongationsphase auf eigene Weise. Kombinierten die Forscher einen CDK9-Blocker mit einem CDK12/13-Blocker, starben die Krebszellen deutlich effizienter als bei einem der einzelnen Wirkstoffe — ein starker Synergieeffekt, den einfache Transkriptionsblocker nicht nachbilden konnten.
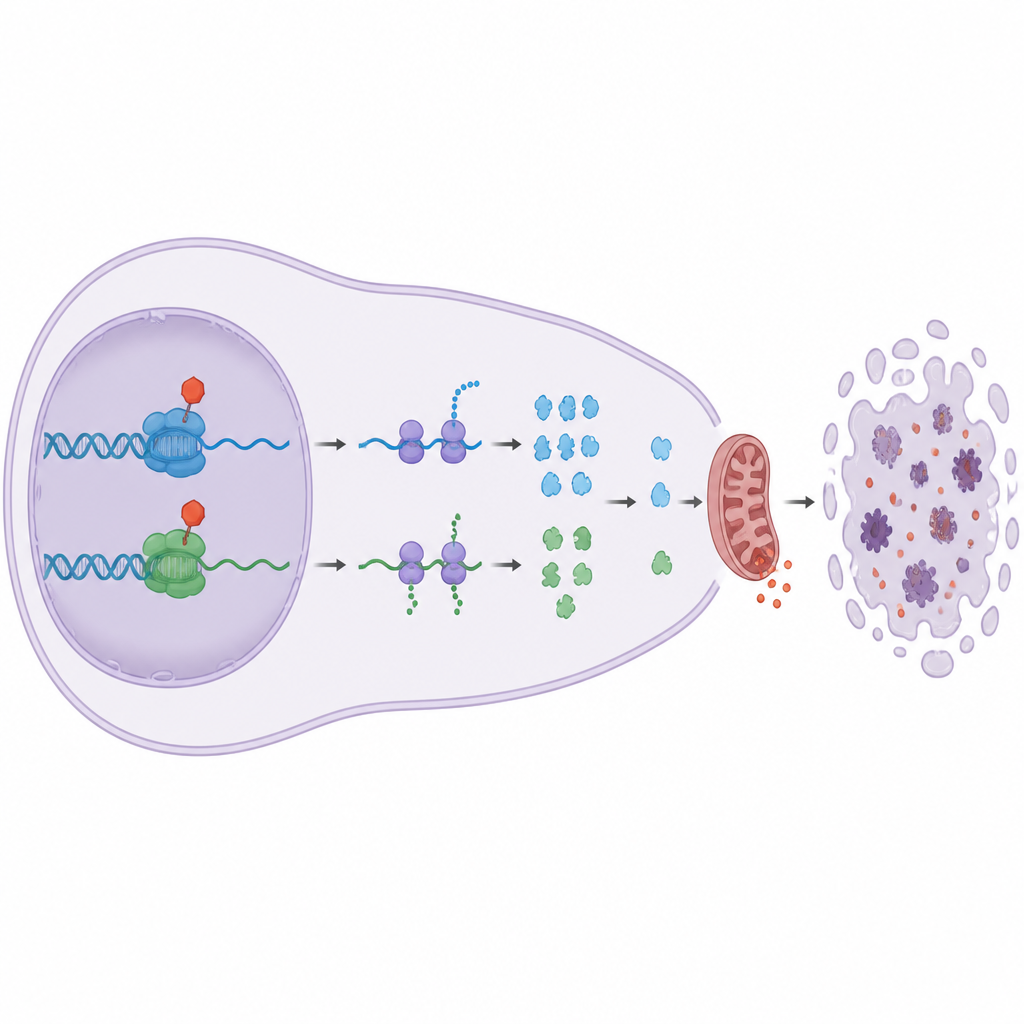
Warum das für die zukünftige Krebsbehandlung wichtig ist
Viele Tumoren sind auf Bcl-2-Familien-„Körperwächter“ angewiesen, um Chemotherapie zu widerstehen; einige überproduzieren sogar Bcl-2 selbst. Diese Studie zeigt, dass das Anvisieren der Elongations-CDKs CDK9 und CDK12/13 hohe Bcl-2-Spiegel umgehen kann, indem Mcl1 und A1 gesenkt werden, die sowohl Bax als auch Bak kontrollieren. Einfach gesagt: Diese Wirkstoffe ziehen mehrere entscheidende Sprossen der Sicherheitsleiter der Krebszelle weg und schonen gleichzeitig andere CDKs, die vorwiegend die normale Zellteilung steuern. Die Arbeit legt nahe, dass fein abgestimmte Inhibitoren von CDK9 und CDK12/13, allein oder in Kombination, eine vielversprechende neue Strategie darstellen könnten, um resistente Blutkrebszellen in die Selbstzerstörung zu treiben und gleichzeitig unerwünschte Schäden im gesunden Gewebe zu minimieren.
Zitation: Krings, K.S., Hatzfeld, J., Weller, S. et al. Inhibition of RNA polymerase II-activating CDK9 and CDK12/13, but not of cell cycle relevant CDKs, induces apoptosis by downregulating the short-lived Bcl-2 proteins Mcl1 and Bfl1/A1. Cell Death Dis 17, 512 (2026). https://doi.org/10.1038/s41419-026-08889-6
Schlüsselwörter: CDK9, CDK12, Mcl1, Apoptose, Blutkrebs