Clear Sky Science · pt
Modelagem DFT baseada em clusters das vibrações Raman em gealcoides amorfos tetraédricos GeS2 e GeSe2
Vidros que moldam a luz
De links de dados de alta velocidade a câmeras infravermelhas, muitos dispositivos fotônicos emergentes dependem de vidros especiais feitos com elementos como enxofre e selênio. Esses materiais “calcogênios” guiam e modulam a luz de maneiras que o vidro comum não consegue. Mas, para ajustar seu desempenho, os cientistas precisam primeiro entender como os átomos se organizam dentro desses sólidos aparentemente desordenados e como essas organizações se manifestam em uma sonda de laboratório simples: o espectro Raman. Este estudo enfrenta esse quebra‑cabeça para vidros e filmes finos de calcogênios à base de germânio, construindo uma ponte entre motivos atômicos invisíveis e as “impressões digitais” vibracionais que os engenheiros realmente medem.
Por que os blocos atômicos importam
Embora os calcogênios amorfos pareçam estruturalmente aleatórios, seus átomos ainda preferem certos arranjos locais. Em vidros baseados em germânio e enxofre ou selênio (GeS2 e GeSe2), o bloco construtivo básico é um pequeno tetraedro: um átomo de germânio rodeado por quatro átomos de calcogênio. Esses tetraedros podem se ligar pelas quinas ou por uma aresta compartilhada, e podem coexistir com características menos regulares, como cadeias de átomos de enxofre ou selênio e ligações entre dois átomos de germânio. A mistura exata desses motivos influencia fortemente a resistência mecânica, a transparência óptica, o índice de refração e a facilidade com que o material muda entre estados amorfo e cristalino — propriedades que sustentam a detecção no infravermelho e circuitos fotônicos de próxima geração.
Usando a computação como um microscópio estrutural
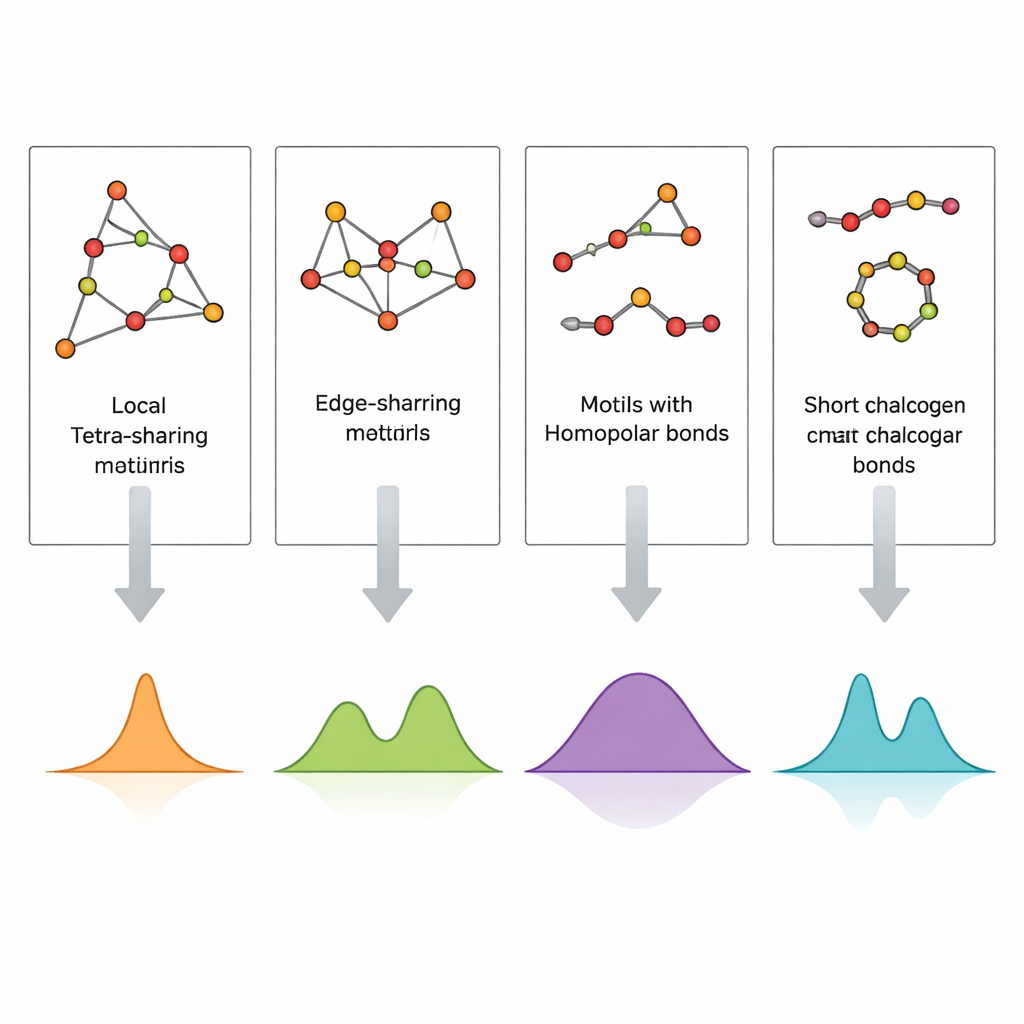
Imagem direta do arranjo dos átomos em um vidro, especialmente em filmes finos de apenas algumas centenas de nanômetros de espessura, é extremamente difícil. A espectroscopia Raman, que registra como a luz é espalhada pelas vibrações atômicas, é muito mais acessível, mas difícil de interpretar porque muitos motivos estruturais podem contribuir para bandas espectrais similares. Os autores enfrentam isso construindo pequenos clusters atômicos cuidadosamente projetados que representam motivos específicos — tetraedros com partilha por quina e por aresta, ligações germânio–germânio e cadeias e anéis curtos de enxofre ou selênio — e então calculando seus espectros vibracionais usando teoria do funcional da densidade (DFT). Ao comparar essas assinaturas Raman simuladas com espectros experimentais tanto de vidros em bloco quanto de filmes finos depositados por sputtering, eles conseguem atribuir picos individuais a estruturas locais concretas.
Casando a teoria com materiais reais
Os espectros calculados reproduzem com notável fidelidade as principais bandas experimentais em GeS2 e GeSe2. Vibrações de tetraedros com partilha por quina explicam os picos mais fortes, enquanto unidades com partilha por aresta dão origem a bandas acompanhantes em frequências ligeiramente mais altas. Sinais de ligações entre dois átomos de germânio e de ligações enxofre–enxofre ou selênio–selênio em cadeias ou anéis curtos aparecem em faixas de frequência bem definidas que coincidem com observações em composições ricas em enxofre ou selênio. Filmes finos exibem características adicionais ou mais intensas vindas de ligações germânio–germânio e bandas mais largas e menos resolvidas, indicando uma maior concentração de defeitos e uma rede mais heterogênea do que em vidros maciços resfriados por fusão. Ao mapear sistematicamente qual cluster produz qual pico, o estudo transforma um espectro Raman complicado em um código estrutural legível.
Olhando além da vizinhança imediata
Para testar se essa abordagem por clusters também captura uma ordem “de alcance médio” mais estendida, a equipe construiu modelos maiores contendo seis tetraedros conectados de maneiras diferentes. Os espectros simulados desses clusters maiores são dominados pelas mesmas vibrações tetraédricas fundamentais dos clusters menores, e as bandas chave permanecem em faixas de frequência estreitas e bem definidas. Isso demonstra que os motivos tetraédricos locais ditam em grande parte a resposta Raman, mesmo quando as redes se tornam mais complexas. Ao mesmo tempo, os cálculos revelam que vibrações envolvendo ligações germânio–germânio são particularmente sensíveis à forma como a rede está conectada, deslocando‑se em frequência conforme as ligações ficam embutidas em ambientes em formato de anel diferentes. Os autores também destacam os limites de seu método — por exemplo, clusters artificialmente lineares podem introduzir deslocamentos artificiais — ressaltando que tais modelos devem ser interpretados com cautela.
Das vibrações a dispositivos melhores
Em termos simples, este trabalho mostra que “maquetes” computacionais bem escolhidas de um vidro podem explicar de forma confiável o que seu espectro Raman revela sobre a estrutura atômica. Para GeS2 e GeSe2, o estudo confirma que unidades tetraédricas e suas conexões formam a espinha dorsal tanto dos vidros em bloco quanto dos filmes finos, enquanto assinaturas específicas revelam onde estão os defeitos e átomos extras de enxofre ou selênio. Com esses vínculos estrutura–vibração em mãos, os pesquisadores podem agora usar medições Raman não apenas como uma ferramenta de diagnóstico, mas como um guia para projetar composições e condições de processamento que produzam vidros e revestimentos calcogênios com comportamento óptico direcionado.
Citação: Halenkovič, T., Němec, P. & Nazabal, V. Cluster-based DFT modeling of Raman vibrations in tetrahedral GeS2 and GeSe2 amorphous chalcogenides. Sci Rep 16, 10009 (2026). https://doi.org/10.1038/s41598-026-40010-x
Palavras-chave: vidro de calcogênio, espectroscopia Raman, teoria do funcional da densidade, filmes finos amorfos, modos vibracionais