Clear Sky Science · fr
Modélisation DFT basée sur des clusters des vibrations Raman dans les chalcogénures amorphes tétraédriques GeS2 et GeSe2
Des verres qui façonnent la lumière
Des liaisons de données à haut débit aux caméras infrarouges, de nombreux dispositifs photoniques émergents reposent sur des verres spéciaux composés d’éléments comme le soufre et le sélénium. Ces matériaux « chalcogénures » guident et modul ent la lumière d’une manière que le verre ordinaire ne permet pas. Mais pour ajuster leurs performances, les chercheurs doivent d’abord comprendre comment les atomes se disposent dans ces solides apparemment désordonnés, et comment ces arrangements se traduisent dans une sonde de laboratoire simple : le spectre Raman. Cette étude s’attaque à ce puzzle pour des verres et des couches minces chalcogénures à base de germanium, en établissant un lien entre motifs atomiques invisibles et « empreintes » vibrationnelles que les ingénieurs mesurent réellement.
Pourquoi les blocs atomiques comptent
Bien que les chalcogénures amorphes paraissent structurellement aléatoires, leurs atomes préfèrent néanmoins certains agencements locaux. Dans les verres à base de germanium et de soufre ou de sélénium (GeS2 et GeSe2), l’unité de base est un petit tétraèdre : un atome de germanium entouré de quatre atomes de chalcogène. Ces tétraèdres peuvent se relier par leurs coins ou partager une arête, et ils peuvent coexister avec des éléments moins réguliers tels que des chaînes de soufre ou de sélénium et des liaisons entre deux atomes de germanium. Le mélange exact de ces motifs influence fortement la résistance mécanique, la transparence optique, l’indice de réfraction et la facilité avec laquelle le matériau bascule entre états amorphe et cristallin — des propriétés essentielles pour la détection infrarouge et les circuits photoniques de nouvelle génération.
Utiliser le calcul comme microscope structural
Imager directement l’arrangement des atomes dans un verre, en particulier dans des couches minces de quelques centaines de nanomètres, est extrêmement difficile. La spectroscopie Raman, qui enregistre comment la lumière est diffusée par les vibrations atomiques, est beaucoup plus accessible mais difficile à interpréter car de nombreux motifs structuraux peuvent contribuer à des bandes spectrales similaires. Les auteurs contournent cette difficulté en construisant de petits clusters atomiques soigneusement conçus représentant des motifs précis — tétraèdres à partage de coin et d’arête, liaisons germanium–germanium, et courtes chaînes ou anneaux de soufre ou de sélénium — puis en calculant leurs spectres vibrationnels par théorie de la fonctionnelle de la densité (DFT). En comparant ces signatures Raman simulées aux spectres expérimentaux issus de verres massifs et de couches minces déposées par pulvérisation, ils peuvent attribuer des pics individuels à des structures locales concrètes.
Faire correspondre la théorie aux matériaux réels
Les spectres calculés reproduisent avec une fidélité remarquable les principales bandes expérimentales de GeS2 et GeSe2. Les vibrations des tétraèdres à partage de coin expliquent les pics les plus intenses, tandis que les unités à partage d’arête donnent lieu à des bandes voisines à des fréquences légèrement plus élevées. Les signaux provenant des liaisons entre deux atomes de germanium, ainsi que des liaisons soufre–soufre ou sélénium–sélénium dans de courtes chaînes ou anneaux, apparaissent dans des fenêtres de fréquence bien définies qui correspondent aux observations dans des compositions riches en soufre ou en sélénium. Les couches minces présentent des caractéristiques supplémentaires ou renforcées liées aux liaisons germanium–germanium et des bandes plus larges et moins résolues, indiquant une concentration plus élevée de défauts et un réseau plus hétérogène que dans les verres massifs refroidis par fusion. En cartographiant systématiquement quel cluster produit quel pic, l’étude transforme un spectre Raman compliqué en un code structural lisible.
Regarder au‑delà du voisinage immédiat
Pour vérifier si cette approche par clusters capture aussi un ordre « à moyenne portée » plus étendu, l’équipe a construit des modèles plus grands contenant six tétraèdres connectés de différentes manières. Les spectres simulés de ces plus grands clusters sont dominés par les mêmes vibrations tétraédriques fondamentales que celles des plus petits, et les bandes clés restent dans des plages de fréquence étroites et bien définies. Cela montre que les motifs tétraédriques locaux dictent en grande partie la réponse Raman, même lorsque les réseaux deviennent plus complexes. En parallèle, les calculs révèlent que les vibrations impliquant des liaisons germanium–germanium sont particulièrement sensibles à la façon dont le réseau est connecté, se déplaçant en fréquence lorsque ces liaisons sont intégrées dans différents environnements en forme d’anneau. Les auteurs soulignent également les limites de leur méthode — par exemple, des clusters linéaires irréalistes peuvent introduire des décalages artificiels — ce qui rappelle que ces modèles doivent être interprétés avec prudence.
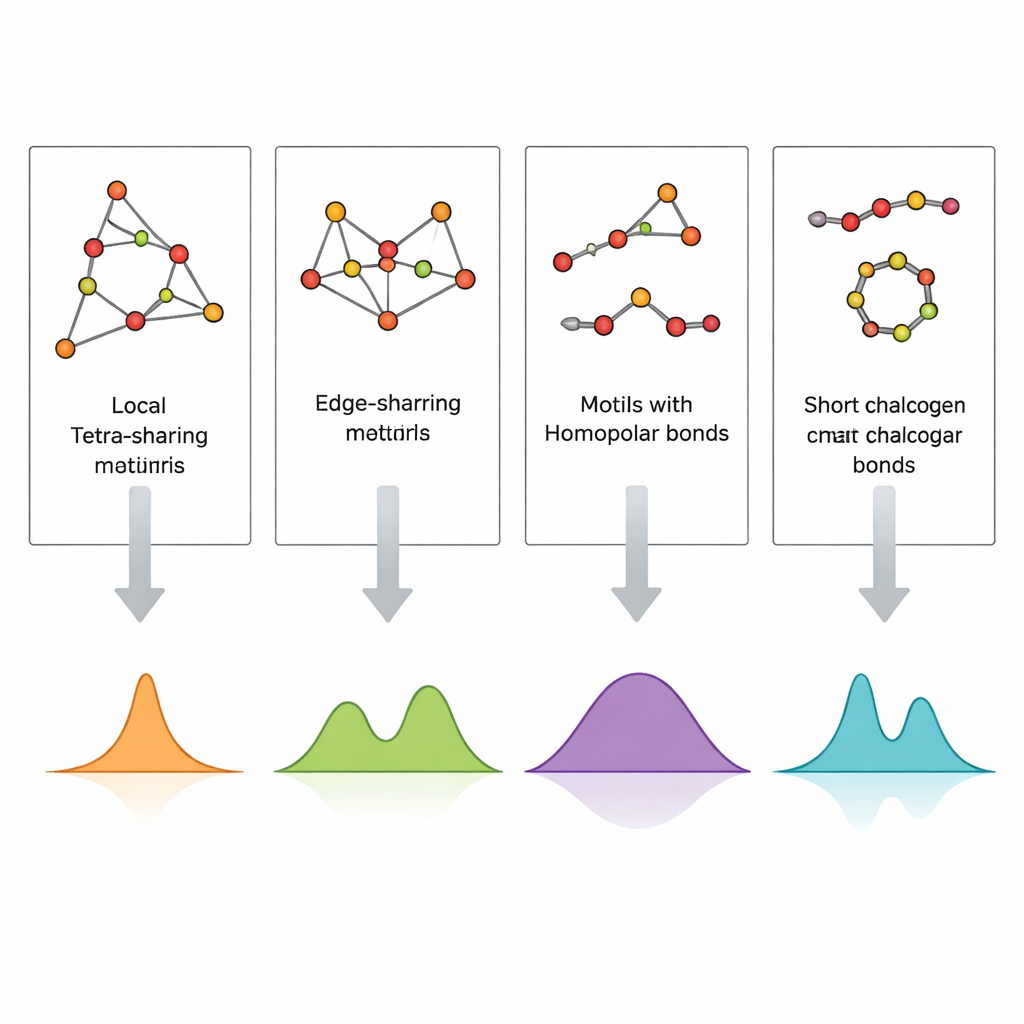
Des vibrations vers de meilleurs dispositifs
En termes simples, ce travail montre que des « maquettes » computationnelles soigneusement choisies d’un verre peuvent expliquer de manière fiable ce que son spectre Raman révèle de sa structure atomique. Pour GeS2 et GeSe2, l’étude confirme que les unités tétraédriques et leurs connexions constituent l’épine dorsale des verres massifs comme des couches minces, tandis que des signatures spécifiques révèlent où se cachent défauts et atomes supplémentaires de soufre ou de sélénium. Avec ces liens structure–vibration en main, les chercheurs peuvent désormais utiliser les mesures Raman non seulement comme outil de diagnostic, mais aussi comme guide pour concevoir des compositions et des conditions de traitement produisant des verres et des revêtements chalcogénures aux comportements optiques ciblés.
Citation: Halenkovič, T., Němec, P. & Nazabal, V. Cluster-based DFT modeling of Raman vibrations in tetrahedral GeS2 and GeSe2 amorphous chalcogenides. Sci Rep 16, 10009 (2026). https://doi.org/10.1038/s41598-026-40010-x
Mots-clés: verre chalcogénure, spectroscopie Raman, théorie de la fonctionnelle de la densité, couches minces amorphes, modes vibrationnels