Clear Sky Science · it
Modellizzazione DFT basata su cluster delle vibrazioni Raman nei chalcogenuri amorfi tetraedrici GeS2 e GeSe2
Vetri che plasmano la luce
Dai collegamenti dati ad alta velocità alle camere a infrarossi, molti dispositivi fotonici emergenti si basano su vetri speciali contenenti elementi come zolfo e selenio. Questi materiali “chalcogenuri” guidano e modulano la luce in modi che il vetro convenzionale non può ottenere. Ma per adattarne le prestazioni, gli scienziati devono prima comprendere come gli atomi si dispongono all’interno di questi solidi apparentemente disordinati e come tali disposizioni si manifestano in una sonda di laboratorio semplice: lo spettro Raman. Questo studio affronta quel problema per vetri e film sottili chalcogenurici a base di germanio, costruendo un ponte tra i motivi atomici invisibili e le “impronte” vibrazionali che gli ingegneri misurano realmente.
Perché contano i mattoni atomici
Sebbene i chalcogenuri amorfi appaiano strutturalmente casuali, gli atomi preferiscono comunque certi arrangiamenti locali. Nei vetri a base di germanio e zolfo o selenio (GeS2 e GeSe2), l’unità fondamentale è un piccolo tetraedro: un atomo di germanio circondato da quattro atomi chalcogenuri. Questi tetraedri possono collegarsi agli spigoli o condividere un bordo, e possono coesistere con elementi meno regolari come catene di atomi di zolfo o selenio e legami tra due atomi di germanio. La miscela esatta di questi motivi influenza fortemente la resistenza meccanica, la trasparenza ottica, l’indice di rifrazione e la facilità con cui il materiale passa dallo stato amorfo a quello cristallino—proprietà che sono alla base del rilevamento nell’infrarosso e dei circuiti fotonici di nuova generazione.
Usare il calcolo come microscopio strutturale
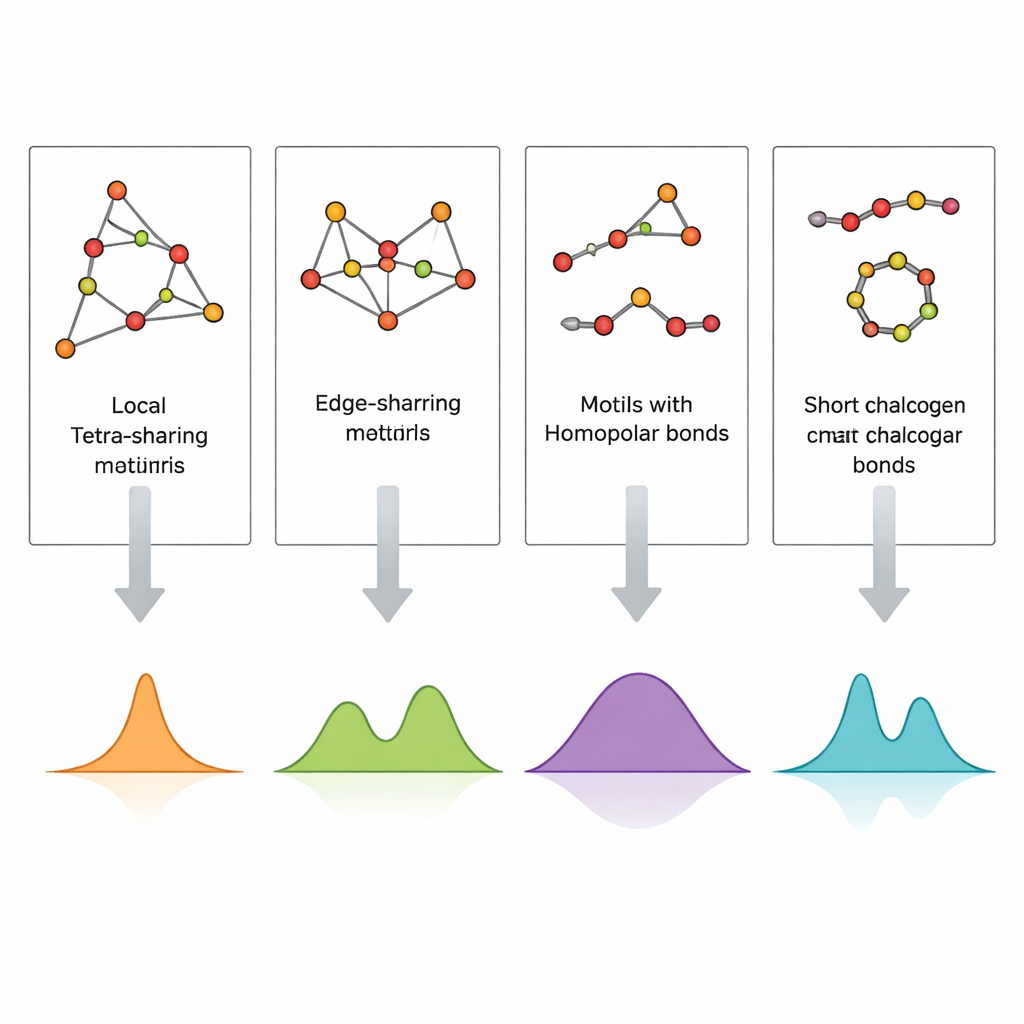
Immaginare direttamente la disposizione degli atomi in un vetro, specialmente in film sottili di poche centinaia di nanometri, è estremamente difficile. La spettroscopia Raman, che registra come la luce viene diffusa dalle vibrazioni atomiche, è molto più accessibile ma difficile da interpretare perché molti motivi strutturali possono contribuire a bande spettrali simili. Gli autori affrontano questo problema costruendo piccoli cluster atomici progettati con cura che rappresentano motivi specifici—tetraedri a condivisione di vertice o di bordo, legami germanio–germanio e brevi catene e anelli di zolfo o selenio—e quindi calcolando i loro spettri vibrazionali mediante teoria del funzionale della densità (DFT). Confrontando queste firme Raman simulate con spettri sperimentali sia di vetri massivi sia di film sputterizzati, possono assegnare picchi individuali a strutture locali concrete.
Far corrispondere la teoria ai materiali reali
Gli spettri calcolati riproducono con notevole fedeltà le principali bande sperimentali in GeS2 e GeSe2. Le vibrazioni dei tetraedri che condividono vertici spiegano i picchi più intensi, mentre le unità a condivisione di bordo generano bande complementari a frequenze leggermente più elevate. I segnali dovuti a legami tra due atomi di germanio e quelli provenienti da legami zolfo–zolfo o selenio–selenio in brevi catene o anelli compaiono in finestre di frequenza ben definite che corrispondono alle osservazioni in composizioni ricche di zolfo o selenio. I film sottili mostrano caratteristiche aggiuntive o più intense dovute ai legami germanio–germanio e bande più ampie e meno risolte, indicando una concentrazione maggiore di difetti e una rete più eterogenea rispetto ai vetri massivi raffreddati per fusione. Mappando sistematicamente quale cluster produce quale picco, lo studio trasforma uno spettro Raman complesso in un codice strutturale leggibile.
Oltre il vicinato immediato
Per verificare se questo approccio a cluster può catturare anche un ordine “a media distanza” più esteso, il gruppo ha costruito modelli più grandi contenenti sei tetraedri connessi in modi diversi. Gli spettri simulati di questi cluster maggiori sono dominati dalle stesse vibrazioni tetraedriche fondamentali dei cluster più piccoli, e le bande chiave restano in intervalli di frequenza ristretti e ben definiti. Ciò dimostra che i motivi tetraedrici locali dettano in larga misura la risposta Raman, anche quando le reti diventano più complesse. Allo stesso tempo, i calcoli rivelano che le vibrazioni che coinvolgono legami germanio–germanio sono particolarmente sensibili al modo in cui la rete è connessa, spostandosi in frequenza quando i legami sono incorporati in ambienti tipo anello differenti. Gli autori evidenziano anche i limiti del metodo—forse cluster eccessivamente lineari e simili a catene possono introdurre spostamenti artificiali—sottolineando che tali modelli devono essere interpretati con cautela.
Dalle vibrazioni a dispositivi migliori
In termini semplici, questo lavoro mostra che “modelli fittizi” computazionali scelti con cura di un vetro possono spiegare in modo affidabile ciò che il suo spettro Raman rivela sulla struttura atomica. Per GeS2 e GeSe2, lo studio conferma che le unità tetraedriche e le loro connessioni costituiscono l’ossatura sia dei vetri massivi sia dei film sottili, mentre firme spettrali specifiche rivelano dove si nascondono difetti e atomi extra di zolfo o selenio. Con questi legami struttura–vibrazione a disposizione, i ricercatori possono ora usare le misure Raman non solo come strumento diagnostico, ma anche come guida per progettare composizioni e condizioni di processo che producano vetri e rivestimenti chalcogenurici con comportamenti ottici mirati.
Citazione: Halenkovič, T., Němec, P. & Nazabal, V. Cluster-based DFT modeling of Raman vibrations in tetrahedral GeS2 and GeSe2 amorphous chalcogenides. Sci Rep 16, 10009 (2026). https://doi.org/10.1038/s41598-026-40010-x
Parole chiave: vetro chalcogenurico, spettroscopia Raman, teoria del funzionale della densità, sottili film amorfi, modi vibrazionali