Clear Sky Science · en
Cluster-based DFT modeling of Raman vibrations in tetrahedral GeS2 and GeSe2 amorphous chalcogenides
Glasses That Shape Light
From high‑speed data links to infrared cameras, many emerging photonic devices rely on special glasses made with elements like sulfur and selenium. These “chalcogenide” materials guide and modulate light in ways ordinary window glass cannot. But to tailor their performance, scientists must first understand how atoms arrange themselves inside these seemingly disordered solids, and how those arrangements show up in a simple laboratory probe: the Raman spectrum. This study tackles that puzzle for germanium‑based chalcogenide glasses and thin films, building a bridge between invisible atomic motifs and the vibrational “fingerprints” that engineers actually measure.
Why Atomic Building Blocks Matter
Although amorphous chalcogenides look structurally random, their atoms still prefer certain local arrangements. In glasses based on germanium and sulfur or selenium (GeS2 and GeSe2), the basic building block is a tiny tetrahedron: one germanium atom surrounded by four chalcogen atoms. These tetrahedra can link at their corners or along a shared edge, and they can coexist with less regular features such as chains of sulfur or selenium atoms and bonds between two germanium atoms. The exact mix of these motifs strongly influences mechanical strength, optical transparency, refractive index, and how easily the material switches between amorphous and crystalline states—properties that underpin infrared sensing and next‑generation photonic circuits.
Using Computation as a Structural Microscope
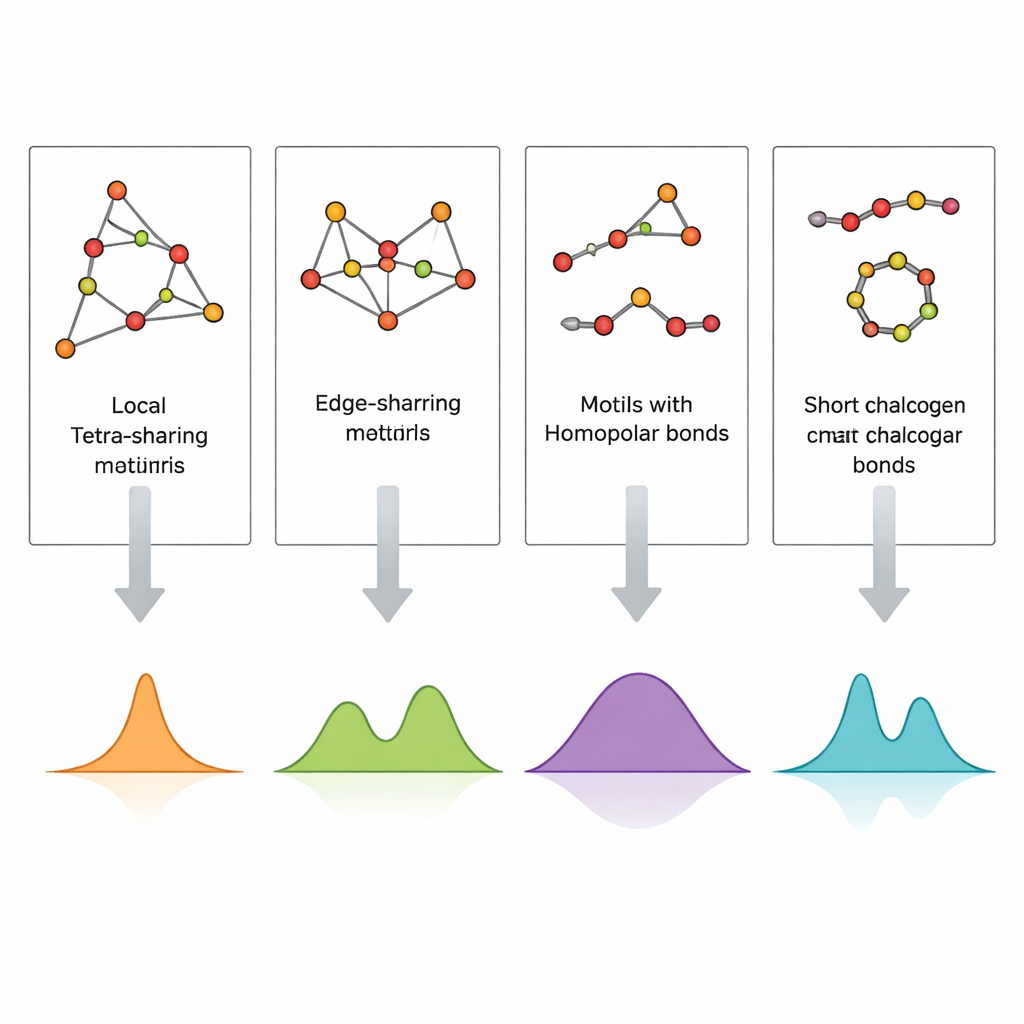
Directly imaging the arrangement of atoms in a glass, especially in thin films only hundreds of nanometers thick, is extremely difficult. Raman spectroscopy, which records how light scatters from atomic vibrations, is far more accessible but hard to interpret because many structural motifs can contribute to similar spectral bands. The authors address this by building small, carefully designed atomic clusters that represent specific motifs—corner‑sharing and edge‑sharing tetrahedra, germanium–germanium bonds, and short sulfur or selenium chains and rings—and then computing their vibrational spectra using density functional theory (DFT). By comparing these simulated Raman signatures with experimental spectra from both bulk glasses and sputtered thin films, they can assign individual peaks to concrete local structures.
Matching Theory to Real Materials
The calculated spectra reproduce the main experimental bands in GeS2 and GeSe2 with striking fidelity. Vibrations of corner‑sharing tetrahedra account for the strongest peaks, while edge‑sharing units give rise to companion bands at slightly higher frequencies. Signals from bonds between two germanium atoms, and from sulfur–sulfur or selenium–selenium links in short chains or rings, appear in well‑defined frequency windows that match observations in sulfur‑ or selenium‑rich compositions. Thin films show additional or stronger features from germanium–germanium bonds and from broader, less resolved bands, indicating a higher concentration of defects and a more heterogeneous network than in melt‑quenched bulk glasses. By systematically mapping which cluster produces which peak, the study turns a complicated Raman spectrum into a readable structural code.
Looking Beyond the Immediate Neighborhood
To test whether this cluster approach can also capture more extended “medium‑range” order, the team constructed larger models containing six tetrahedra connected in different ways. The simulated spectra of these bigger clusters are dominated by the same fundamental tetrahedral vibrations as the smaller ones, and the key bands remain in narrow, well‑defined frequency ranges. This shows that the local tetrahedral motifs largely dictate the Raman response, even as networks grow more complex. At the same time, the calculations reveal that vibrations involving germanium–germanium bonds are particularly sensitive to how the network is connected, shifting in frequency as bonds are embedded in different ring‑like environments. The authors also highlight the limits of their method—for example, unrealistically straight, chain‑like clusters can introduce artificial shifts—underscoring that such models must be interpreted with care.
From Vibrations to Better Devices
In plain terms, this work shows that carefully chosen computational “mock‑ups” of a glass can reliably explain what its Raman spectrum is telling us about atomic structure. For GeS2 and GeSe2, the study confirms that tetrahedral units and their connections form the backbone of both bulk glasses and thin films, while specific signatures reveal where defects and extra sulfur or selenium atoms hide. With these structure‑vibration links in hand, researchers can now use Raman measurements not just as a diagnostic tool, but as a guide for designing compositions and processing conditions that yield chalcogenide glasses and coatings with targeted optical behavior.
Citation: Halenkovič, T., Němec, P. & Nazabal, V. Cluster-based DFT modeling of Raman vibrations in tetrahedral GeS2 and GeSe2 amorphous chalcogenides. Sci Rep 16, 10009 (2026). https://doi.org/10.1038/s41598-026-40010-x
Keywords: chalcogenide glass, Raman spectroscopy, density functional theory, amorphous thin films, vibrational modes