Clear Sky Science · nl
Clustergebaseerde DFT‑modellering van Raman‑vibraties in tetraëdrische GeS2 en GeSe2 amorfe chalcogeniden
Glazen die licht vormen
Van hogesnelheidsdataverbindingen tot infraroodcamera’s: veel nieuwe fotonische apparaten vertrouwen op speciale glazen gemaakt met elementen zoals zwavel en selenium. Deze “chalcogenide” materialen geleiden en moduleren licht op manieren die gewoon vensterglas niet kan. Om hun prestaties te optimaliseren, moeten wetenschappers eerst begrijpen hoe atomen zich in deze ogenschijnlijk wanordelijke vaste stoffen schikken, en hoe die ordening naar voren komt in een eenvoudig laboratoriuminstrument: het Raman‑spectrum. Deze studie pakt die puzzel aan voor germanium‑gebaseerde chalcogenideglazen en dunne films en bouwt een brug tussen onzichtbare atomaire motieven en de trillings‑“vingerafdrukken” die ingenieurs meten.
Waarom atomaire bouwstenen ertoe doen
Hoewel amorfe chalcogeniden er structureel willekeurig uitzien, geven hun atomen toch de voorkeur aan bepaalde lokale ordeningen. In glazen op basis van germanium en zwavel of selenium (GeS2 en GeSe2) is het basiselement een klein tetraëder: één germaniumatoom omringd door vier chalcogenen. Deze tetraëders kunnen elkaar ontmoeten via hoekdeling of door een gedeelde rand, en ze kunnen naast minder regelmatige kenmerken voorkomen zoals ketens van zwavel‑ of seleniumatomen en bindingen tussen twee germaniumatomen. De precieze samenstelling van deze motieven beïnvloedt sterk de mechanische sterkte, optische transparantie, de brekingsindex en hoe gemakkelijk het materiaal tussen amorfe en kristallijne toestanden schakelt—eigenschappen die ten grondslag liggen aan infraroodsensing en fotonische schakelingen van de volgende generatie.
Computatie als structurele microscoop
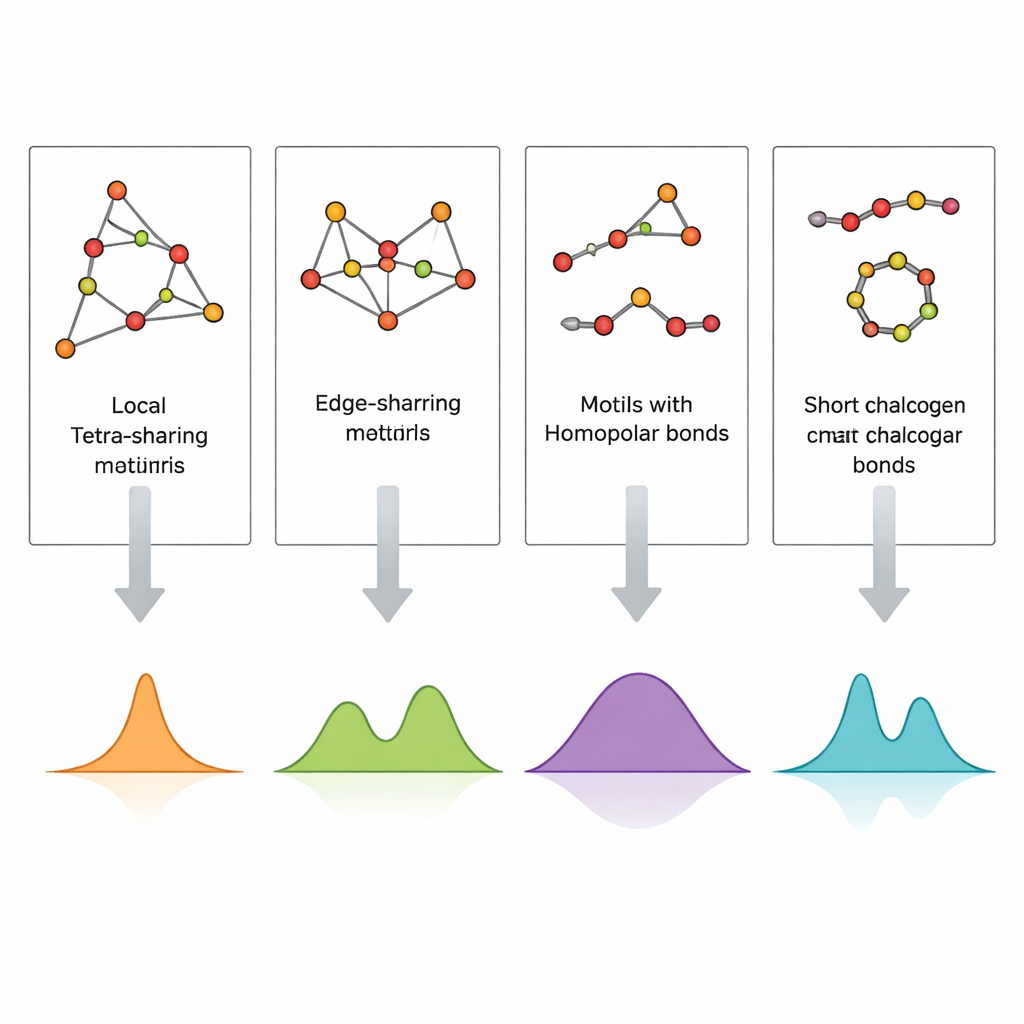
De ordening van atomen in een glas rechtstreeks in beeld brengen, vooral in dunne films van slechts enkele honderden nanometers, is extreem moeilijk. Raman‑spectroscopie, die vastlegt hoe licht verstrooit door atomische vibraties, is veel toegankelijker maar lastig te interpreteren omdat veel structurele motieven aan vergelijkbare spectrale banden kunnen bijdragen. De auteurs pakken dit aan door kleine, zorgvuldig ontworpen atomaire clusters te bouwen die specifieke motieven vertegenwoordigen—hoekdelende en randdelende tetraëders, germanium–germaniumbindingen en korte zwavel‑ of seleniumketens en ringen—en vervolgens hun vibratiespectra te berekenen met dichtheidsfunctionaaltheorie (DFT). Door deze gesimuleerde Raman‑handtekeningen te vergelijken met experimentele spectra van zowel bulkglazen als gesputterde dunne films, kunnen ze individuele pieken toewijzen aan concrete lokale structuren.
Theorie afstemmen op echte materialen
De berekende spectra reproduceren de belangrijkste experimentele banden in GeS2 en GeSe2 met opvallende nauwkeurigheid. Trillingen van hoekdelende tetraëders verklaren de sterkste pieken, terwijl randdelende eenheden begeleidende banden veroorzaken op iets hogere frequenties. Signalen van bindingen tussen twee germaniumatomen, en van zwavel‑zwavel of selenium‑selenium verbindingen in korte ketens of ringen, verschijnen in duidelijk afgebakende frequentievensters die overeenkomen met waarnemingen in zwavel‑ of seleniumrijke samenstellingen. Dunne films vertonen aanvullende of sterkere kenmerken van germanium–germaniumbindingen en van bredere, minder opgeloste banden, wat wijst op een hogere concentratie defecten en een heterogener netwerk dan in smelt‑gequenched bulkglazen. Door systematisch in kaart te brengen welke cluster welke piek produceert, verandert de studie een complex Raman‑spectrum in een leesbare structurele code.
Kijken voorbij de directe buurt
Om te testen of deze clusterbenadering ook meer uitgebreide “medium‑range” orde kan vastleggen, bouwde het team grotere modellen die zes tetraëders bevatten die op verschillende manieren verbonden zijn. De gesimuleerde spectra van deze grotere clusters worden gedomineerd door dezelfde fundamentele tetraëdrale vibraties als de kleinere modellen, en de sleutelbanden blijven in smalle, goed gedefinieerde frequentiegebieden. Dit toont aan dat de lokale tetraëdrale motieven grotendeels de Raman‑respons bepalen, zelfs wanneer netwerken complexer worden. Tegelijkertijd laten de berekeningen zien dat vibraties die germanium–germaniumbindingen omvatten bijzonder gevoelig zijn voor hoe het netwerk is verbonden, en in frequentie verschuiven naarmate bindingen in verschillende ringachtige omgevingen ingebed zitten. De auteurs benadrukken ook de beperkingen van hun methode—bijvoorbeeld onrealistisch rechte, ketenachtige clusters kunnen kunstmatige verschuivingen introduceren—en benadrukken dat dergelijke modellen met voorzichtigheid geïnterpreteerd moeten worden.
Van vibraties naar betere apparaten
Kort gezegd toont dit werk aan dat zorgvuldig gekozen computationele “mock‑ups” van een glas betrouwbaar kunnen verklaren wat het Raman‑spectrum ons vertelt over de atomaire structuur. Voor GeS2 en GeSe2 bevestigt de studie dat tetraëdrale eenheden en hun verbindingen de ruggengraat vormen van zowel bulkglazen als dunne films, terwijl specifieke signaturen onthullen waar defecten en extra zwavel‑ of seleniumatomen zich bevinden. Met deze koppelingen tussen structuur en vibratie kunnen onderzoekers Raman‑metingen nu niet alleen als diagnostisch hulpmiddel gebruiken, maar ook als leidraad voor het ontwerpen van samenstellingen en verwerkingscondities die chalcogenideglazen en‑coatings opleveren met doelgerichte optische eigenschappen.
Bronvermelding: Halenkovič, T., Němec, P. & Nazabal, V. Cluster-based DFT modeling of Raman vibrations in tetrahedral GeS2 and GeSe2 amorphous chalcogenides. Sci Rep 16, 10009 (2026). https://doi.org/10.1038/s41598-026-40010-x
Trefwoorden: chalcogenideglas, Raman‑spectroscopie, dichtheidsfunctionaaltheorie, amorfe dunne films, vibratiemodi