Clear Sky Science · de
Clusterbasierte DFT-Modellierung der Raman‑Vibrationen in tetraedrischem GeS2 und GeSe2 amorphen Chalkogeniden
Gläser, die das Licht formen
Von Hochgeschwindigkeitsdatenverbindungen bis zu Infrarotkameras beruhen viele neuartige photonische Bauteile auf speziellen Gläsern, die Elemente wie Schwefel und Selen enthalten. Diese „Chalkogenid“-Materialien leiten und modulieren Licht auf Weisen, die gewöhnliches Fensterglas nicht ermöglicht. Um ihre Eigenschaften gezielt zu gestalten, müssen Forschende jedoch zunächst verstehen, wie sich die Atome in diesen scheinbar ungeordneten Festkörpern anordnen und wie sich diese Anordnungen in einer einfachen Laboruntersuchung zeigen: dem Raman‑Spektrum. Die vorliegende Studie geht dieses Rätsel für germaniumbasierte Chalkogenidgläser und Dünnschichten an und schlägt eine Brücke zwischen unsichtbaren atomaren Motiven und den vibrationalen „Fingerabdrücken“, die Ingenieure tatsächlich messen.
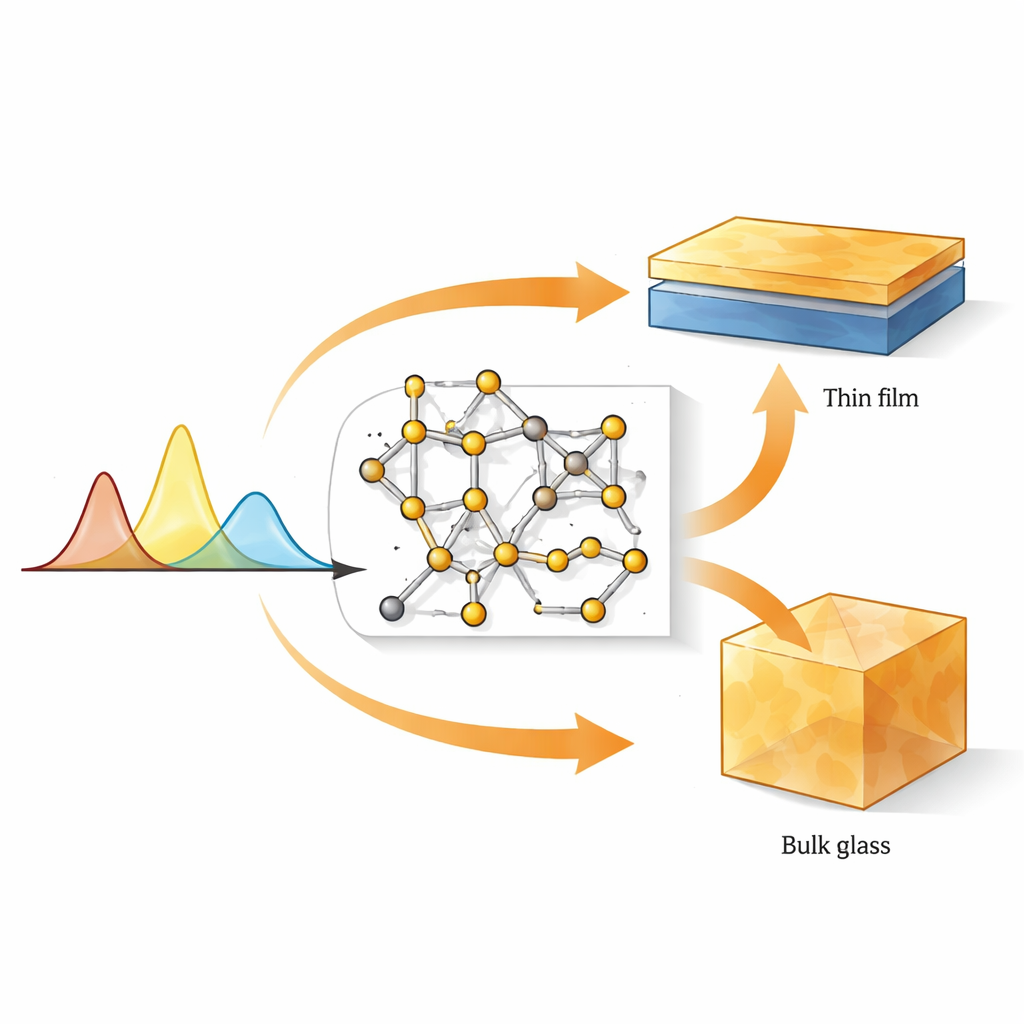
Warum atomare Bausteine zählen
Obwohl amorphe Chalkogenide strukturell zufällig wirken, bevorzugen ihre Atome dennoch bestimmte lokale Anordnungen. In Gläsern auf Basis von Germanium und Schwefel oder Selen (GeS2 und GeSe2) ist das grundlegende Bauelement ein kleines Tetraeder: ein Germaniumatom, umgeben von vier Chalkogenatomen. Diese Tetraeder können an ihren Ecken oder entlang einer gemeinsamen Kante verknüpft sein und koexistieren mit weniger regelmäßigen Merkmalen wie Ketten aus Schwefel‑ oder Selenatomen sowie Bindungen zwischen zwei Germaniumatomen. Die genaue Mischung dieser Motive beeinflusst stark mechanische Festigkeit, optische Transparenz, Brechungsindex und die Leichtigkeit, mit der das Material zwischen amorphem und kristallinem Zustand wechselt – Eigenschaften, die der Infrarotdetektion und photonischen Schaltkreisen der nächsten Generation zugrunde liegen.
Rechnen als strukturelles Mikroskop
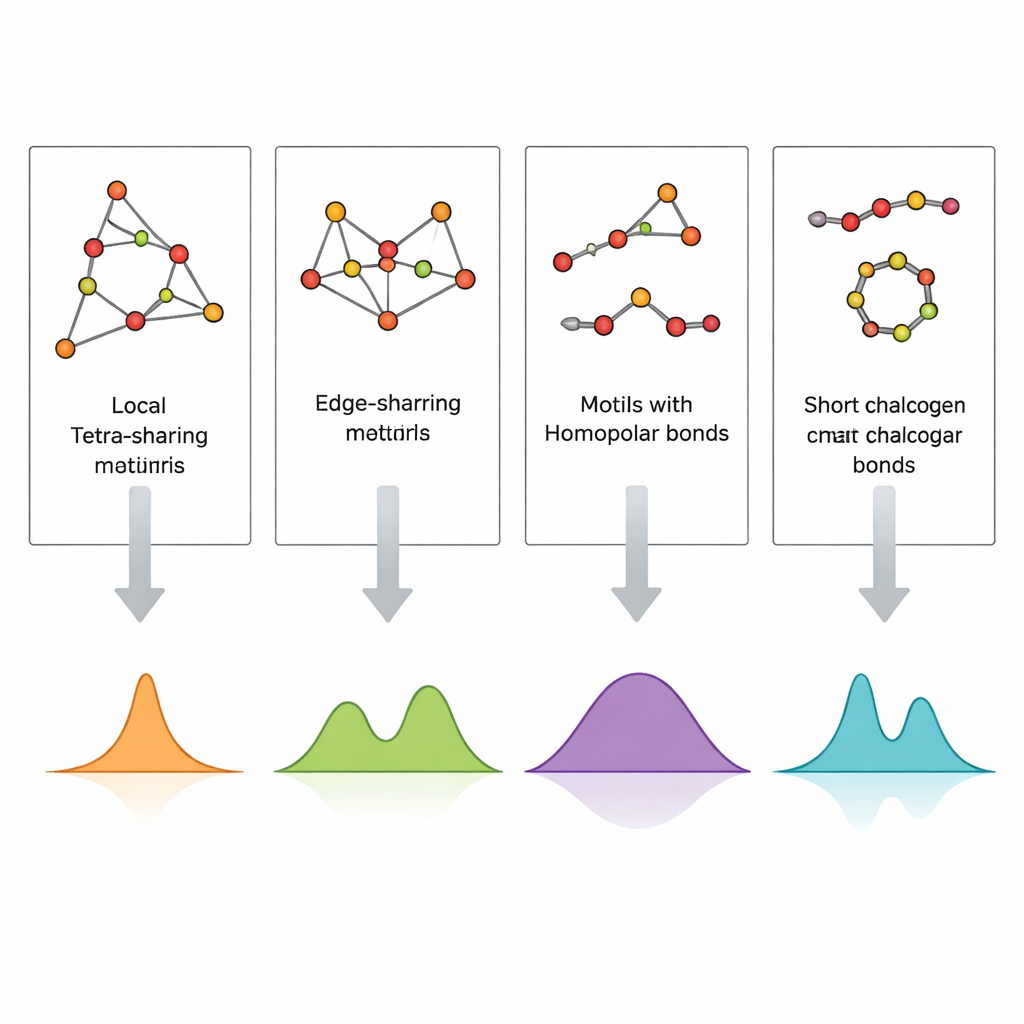
Die direkte Abbildung der Atomordnung in einem Glas, besonders in Dünnschichten von nur einigen hundert Nanometern Dicke, ist extrem schwierig. Die Ramanspektroskopie, die aufzeichnet, wie Licht an atomaren Schwingungen gestreut wird, ist viel zugänglicher, aber schwer zu interpretieren, weil viele Strukturmotive zu ähnlichen Spektralbändern beitragen können. Die Autoren begegnen diesem Problem, indem sie kleine, sorgfältig gestaltete atomare Cluster bauen, die spezifische Motive repräsentieren – eckverknüpfte und kantenverknüpfte Tetraeder, Germanium‑Germanium‑Bindungen sowie kurze Schwefel‑ oder Selenketten und Ringe – und anschließend deren Vibrationsspektren mit Dichtematrix‑Funktionaltheorie (DFT) berechnen. Durch den Vergleich dieser simulierten Raman‑Signaturen mit experimentellen Spektren sowohl aus Volumengläsern als auch aus gesputterten Dünnschichten können sie einzelne Peaks konkreten lokalen Strukturen zuordnen.
Theorie an reale Materialien anpassen
Die berechneten Spektren reproduzieren die wichtigsten experimentellen Banden in GeS2 und GeSe2 mit auffälliger Treue. Schwingungen eckverknüpfter Tetraeder erklären die stärksten Peaks, während kantenverknüpfte Einheiten Gefährtenbänder bei leicht höheren Frequenzen hervorrufen. Signale von Bindungen zwischen zwei Germaniumatomen sowie von Schwefel‑Schwefel‑ oder Selen‑Selen‑Verknüpfungen in kurzen Ketten oder Ringen erscheinen in gut definierten Frequenzfenstern, die zu Beobachtungen in schwefel‑ bzw. selenreichen Zusammensetzungen passen. Dünnschichten zeigen zusätzliche oder verstärkte Merkmale durch Germanium‑Germanium‑Bindungen und durch breitere, weniger aufgelöste Bänder, was auf eine höhere Defektdichte und ein heterogeneres Netzwerk im Vergleich zu schmelzgequenchten Volumengläsern hinweist. Durch das systematische Kartieren, welches Cluster welchen Peak erzeugt, verwandelt die Studie ein kompliziertes Raman‑Spektrum in einen lesbaren Strukturcode.
Über die unmittelbare Nachbarschaft hinaus blicken
Um zu prüfen, ob dieser Clusteransatz auch erweitert‑räumliche „mittelräumliche“ Ordnung erfassen kann, konstruierte das Team größere Modelle mit sechs Tetraedern, die auf verschiedene Weise verbunden sind. Die simulierten Spektren dieser größeren Cluster werden von denselben fundamentalen tetraedrischen Schwingungen dominiert wie die kleineren, und die Schlüsselbänder bleiben in engen, gut definierten Frequenzbereichen. Das zeigt, dass die lokalen tetraedrischen Motive die Raman‑Antwort weitgehend bestimmen, selbst wenn Netzwerke komplexer werden. Gleichzeitig offenbaren die Berechnungen, dass Schwingungen, die Germanium‑Germanium‑Bindungen betreffen, besonders empfindlich auf die Netzwerk‑Topologie reagieren und in der Frequenz verschoben werden, wenn Bindungen in unterschiedliche ringähnliche Umgebungen eingebettet sind. Die Autoren heben auch die Grenzen ihrer Methode hervor – etwa können unrealistisch gerade, kettenartige Cluster künstliche Verschiebungen einführen – und betonen, dass solche Modelle mit Vorsicht interpretiert werden müssen.
Von Schwingungen zu besseren Bauteilen
Kurz gesagt zeigt diese Arbeit, dass sorgfältig ausgewählte rechnerische „Mock‑ups“ eines Glases zuverlässig erklären können, was dessen Raman‑Spektrum über die atomare Struktur aussagt. Für GeS2 und GeSe2 bestätigt die Studie, dass tetraedrische Einheiten und ihre Verknüpfungen das Rückgrat sowohl von Volumengläsern als auch von Dünnschichten bilden, während spezifische Signaturen verraten, wo Defekte und zusätzliche Schwefel‑ oder Selenatome verborgen sind. Mit diesen Struktur‑Schwingungs‑Verknüpfungen können Forschende Raman‑Messungen nun nicht nur als Diagnosewerkzeug nutzen, sondern als Leitfaden zur Gestaltung von Zusammensetzungen und Verarbeitungsbedingungen, die Chalkogenidgläser und Beschichtungen mit gezieltem optischem Verhalten liefern.
Zitation: Halenkovič, T., Němec, P. & Nazabal, V. Cluster-based DFT modeling of Raman vibrations in tetrahedral GeS2 and GeSe2 amorphous chalcogenides. Sci Rep 16, 10009 (2026). https://doi.org/10.1038/s41598-026-40010-x
Schlüsselwörter: Chalkogenidglas, Ramanspektroskopie, Dichtematrixfunktionaltheorie, amorphe Dünnschichten, Schwingungsmoden