Clear Sky Science · pt
Uma abordagem imparcial para medir alterações aberrantes na metilação do DNA
Por que pequenas marcas químicas no DNA importam
Nossas células carregam milhões de pequenas marcas químicas no DNA que ajudam a controlar quais genes estão ligados ou desligados. No câncer e em outras doenças, essas marcas podem mudar de forma prejudicial. Este estudo pergunta algo simples, mas importante: estamos medindo essas mudanças da maneira correta ou uma régua comum tem ocultado silenciosamente alguns dos sinais de alerta mais relevantes?
Como os cientistas geralmente acompanham as marcas no DNA
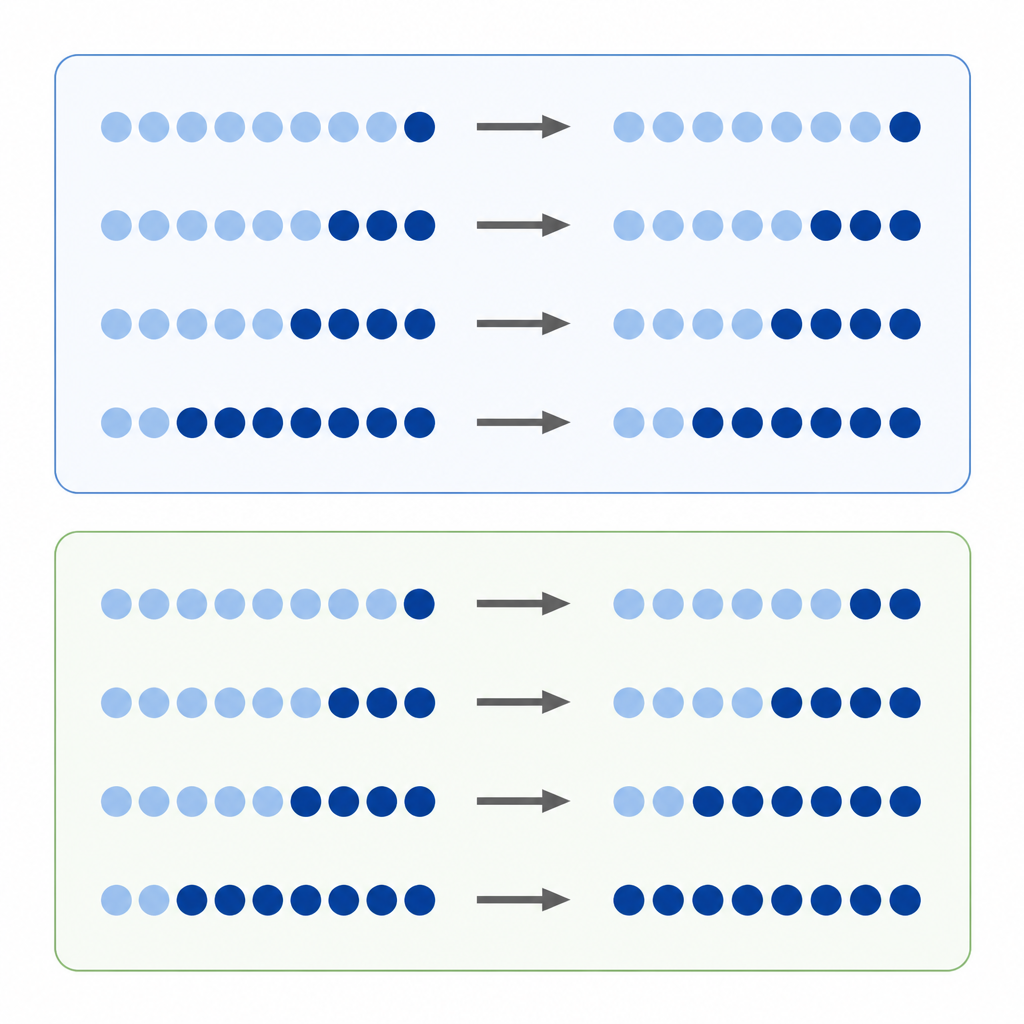
A metilação do DNA é uma das marcas químicas mais conhecidas em nossos genes. Pesquisadores frequentemente comparam quanta metilação um sítio de DNA apresenta em um tumor versus tecido normal, tratando a diferença direta nos níveis como uma medida da gravidade da alteração. Essa abordagem de “diferença absoluta” parece natural, mas os níveis de metilação estão limitados entre um mínimo e um máximo, e cada sítio tende a se situar em um nível inicial preferencial. Isso significa que um sítio que começa altamente metilado tem mais margem para perder marcas do que um sítio que parte de um nível baixo, o que pode distorcer a percepção de quais mudanças parecem grandes ou pequenas.
Uma nova maneira de pensar sobre mudança
Os autores propõem que muitas alterações na metilação se comportam mais como variações percentuais do que como ganhos ou perdas simples. Em vez de perguntar “quantos pontos este sítio se moveu”, eles perguntam “qual foi o tamanho da mudança em comparação com o ponto de partida?”. Eles chamam isso de mudança relativa. Se cada sítio tende a ganhar ou perder metilação a uma taxa percentual aproximadamente igual, então focar apenas em diferenças absolutas favorecerá alguns sítios e ignorará outros, criando um viés oculto na forma como interpretamos o epigenoma do câncer.
Testando a ideia em células e tumores
Para investigar essa hipótese, a equipe analisou dados de culturas celulares tratadas com uma droga que remove metilação e de milhares de amostras tumorais representando diversos tipos de câncer. Em células tratadas, sítios que começaram com alta metilação exibiram as maiores quedas absolutas, como esperado. Mas quando os pesquisadores observaram as mudanças relativas, viram que sítios ao longo de toda a faixa de metilação perderam uma fração semelhante de suas marcas originais. Em grandes conjuntos de dados de câncer, o mesmo padrão apareceu: enquanto as mudanças absolutas ainda dependiam fortemente dos níveis iniciais, as mudanças relativas se alinharam para revelar variações percentuais semelhantes entre diferentes sítios e tipos tumorais. Simulações computacionais ajudaram a mostrar que esses padrões eram improváveis de serem resultado de ruído aleatório.
Encontrando sinais de câncer mais claros
Os autores então perguntaram qual medida faz um trabalho melhor em destacar mudanças biologicamente relevantes. Eles compararam quão bem diferenças absolutas e relativas detectavam “assinaturas” conhecidas de metilação ligadas a fatores como idade, tabagismo e um padrão específico no câncer colorretal. As mudanças relativas foram mais sensíveis nos extremos, como sítios que eram quase sempre metilados ou quase nunca metilados em tecido saudável. Usar variações relativas também revelou genes envolvidos em adesão celular, metabolismo, sinalização e atividade imune — processos intimamente ligados ao crescimento e à disseminação tumoral. Em contraste, confiar apenas em diferenças absolutas muitas vezes apontava para genes envolvidos em vias relacionadas ao cérebro, que são mais difíceis de conectar diretamente ao comportamento do câncer.
Por que essa nova visão importa
Ao tratar as mudanças na metilação como variações relativas ao ponto de partida de cada sítio, o estudo oferece uma lente menos tendenciosa sobre o genoma tumoral. Essa perspectiva captura sinais importantes em regiões que métodos padrão tendem a perder, especialmente onde a perda de metilação pode desestabilizar cromossomos ou ativar elementos de DNA silenciosos. O trabalho sugere que grande parte do que considerávamos as maiores alterações de metilação no câncer pode refletir nossa forma de medir, e não a biologia em si.
O que isso significa para pesquisas futuras
Para não especialistas, a conclusão é que a maneira como medimos a mudança pode alterar dramaticamente as histórias que contamos sobre doenças. Este artigo argumenta que a metilação do DNA no câncer costuma mudar por uma porcentagem relativamente consistente em grande parte do genoma, e que focar na mudança relativa ajuda a revelar vias mais claramente ligadas ao crescimento e à disseminação tumoral. Adotar essa nova abordagem pode afiar esforços futuros para usar padrões de metilação na compreensão do risco de câncer, no acompanhamento da evolução tumoral e, talvez, na orientação de diagnóstico e tratamento, sem alterar os dados subjacentes.
Citação: Downs, B.M., Hu, J., Park, J.S. et al. An unbiased approach to measure aberrant DNA methylation alterations. Nat Commun 17, 4522 (2026). https://doi.org/10.1038/s41467-026-71089-5
Palavras-chave: metilação do DNA, epigenética do câncer, biomarcadores epigenéticos, regulação do genoma, análise de metilação