Clear Sky Science · fr
Une approche impartiale pour mesurer les altérations aberrantes de la méthylation de l’ADN
Pourquoi de petites étiquettes chimiques sur l’ADN comptent
Nos cellules portent des millions de petites étiquettes chimiques sur leur ADN qui contribuent à contrôler quels gènes sont activés ou désactivés. Dans le cancer et d’autres maladies, ces étiquettes peuvent se modifier de façon nocive. Cette étude pose une question simple mais importante : mesurons‑nous ces changements de la bonne manière, ou un étalon commun a‑t‑il discrètement caché certains des signaux d’alerte les plus importants ?
Comment les scientifiques suivent habituellement le marquage de l’ADN
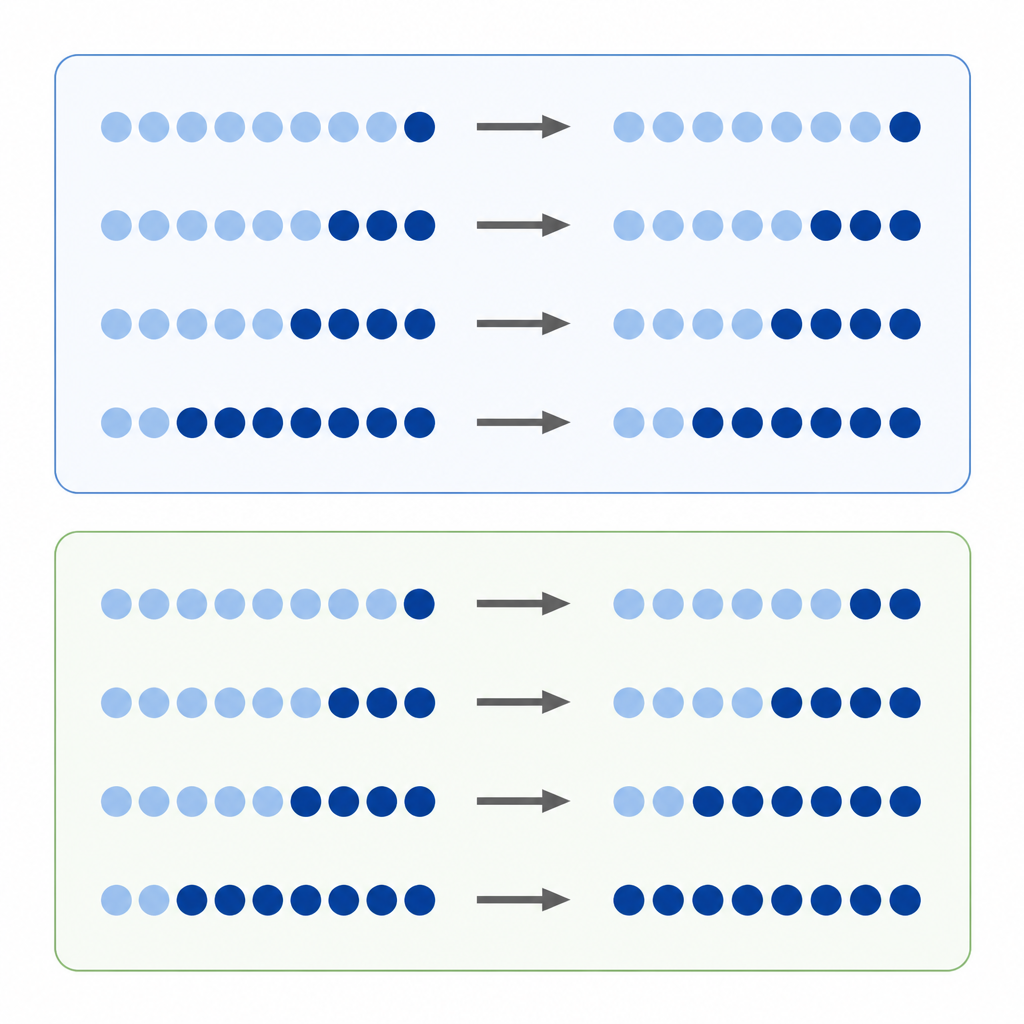
La méthylation de l’ADN est l’une des étiquettes chimiques les mieux connues sur nos gènes. Les chercheurs comparent souvent le degré de méthylation d’un site d’ADN dans une tumeur par rapport au tissu normal, en traitant la différence directe des niveaux comme une mesure de la gravité du changement. Cette approche de « différence absolue » semble naturelle, mais les niveaux de méthylation sont limités entre une valeur minimale et maximale, et chaque site tend à se situer à un niveau de départ préféré. Cela signifie qu’un site très méthylé au départ a plus de marge pour perdre des marques qu’un site peu méthylé, ce qui peut fausser l’apparence de l’amplitude des changements.
Une nouvelle façon de penser le changement
Les auteurs proposent que de nombreux changements de méthylation se comportent davantage comme des variations en pourcentage que comme de simples gains ou pertes. Plutôt que de demander « de combien de points ce site a‑t‑il bougé », ils demandent « quelle a été l’importance du changement par rapport à son point de départ ? » Ils appellent cela un changement relatif. Si chaque site tend à gagner ou perdre de la méthylation à peu près au même taux en pourcentage, se concentrer uniquement sur les différences absolues favorisera certains sites et en négligera d’autres, créant un biais caché dans notre lecture de l’épigénome tumoral.
Tester l’idée dans des cellules et des tumeurs
Pour explorer cette idée, l’équipe a analysé des données de cultures cellulaires traitées par un médicament qui élimine la méthylation et des milliers d’échantillons tumoraux représentant de nombreux types de cancer. Dans les cellules traitées, les sites qui commençaient avec une forte méthylation ont montré les plus grosses chutes absolues, comme prévu. Mais quand les chercheurs ont regardé les changements relatifs, ils ont observé que des sites couvrant toute la gamme de méthylation perdaient une fraction similaire de leurs marques d’origine. Dans de larges jeux de données tumorales, le même schéma est apparu : alors que les changements absolus dépendaient fortement du niveau de départ, les changements relatifs mettaient en évidence des variations en pourcentage similaires entre différents sites et types de tumeurs. Des simulations informatiques ont aidé à montrer que ces motifs étaient peu susceptibles de provenir du bruit aléatoire.
Trouver des signaux cancéreux plus nets
Les auteurs ont ensuite demandé quelle mesure mettait le mieux en évidence les changements biologiquement significatifs. Ils ont comparé la capacité des changements absolus et relatifs à détecter des « signatures » de méthylation connues liées à des facteurs comme l’âge, le tabagisme et un motif particulier du cancer colorectal. Les changements relatifs étaient plus sensibles aux extrêmes, comme les sites presque toujours méthylés ou presque jamais méthylés dans les tissus sains. L’utilisation des variations relatives a également révélé des gènes impliqués dans l’adhésion cellulaire, le métabolisme, la signalisation et l’activité immunitaire, tous des processus étroitement liés à la croissance et à la propagation tumorales. En revanche, s’en tenir aux différences absolues pointait souvent vers des gènes impliqués dans des voies cérébrales, plus difficiles à relier directement au comportement du cancer.
Pourquoi cette nouvelle vision importe
En traitant les variations de méthylation comme des changements relatifs à l’état initial de chaque site, l’étude offre un regard moins biaisé sur le génome tumoral. Cette perspective capture des signaux importants dans des régions que les méthodes standard ont tendance à manquer, en particulier là où la perte de méthylation peut déstabiliser les chromosomes ou réveiller des éléments d’ADN silencieux. Le travail suggère qu’une grande partie de ce que nous avons considéré comme les plus grands changements de méthylation dans le cancer peut refléter notre étalon de mesure, et non la biologie elle‑même.
Ce que cela implique pour la recherche future
Pour les non‑spécialistes, l’essentiel est que la façon dont nous mesurons le changement peut modifier radicalement les récits que nous construisons sur la maladie. Cet article soutient que la méthylation de l’ADN dans le cancer varie généralement d’un pourcentage assez constant sur une grande partie du génome, et que se concentrer sur le changement relatif aide à révéler plus clairement les voies liées à la croissance et à la dissémination tumorales. Adopter cette nouvelle approche pourrait affiner les efforts futurs visant à utiliser les motifs de méthylation pour comprendre le risque de cancer, suivre l’évolution tumorale et, peut‑être, orienter le diagnostic et le traitement, sans modifier du tout les données sous‑jacentes.
Citation: Downs, B.M., Hu, J., Park, J.S. et al. An unbiased approach to measure aberrant DNA methylation alterations. Nat Commun 17, 4522 (2026). https://doi.org/10.1038/s41467-026-71089-5
Mots-clés: Méthylation de l’ADN, épigénétique du cancer, biomarqueurs épigénétiques, régulation du génome, analyse de la méthylation