Clear Sky Science · en
An unbiased approach to measure aberrant DNA methylation alterations
Why tiny chemical tags on DNA matter
Our cells carry millions of small chemical tags on their DNA that help control which genes are on or off. In cancer and other diseases, these tags can shift in harmful ways. This study asks a simple but important question: are we measuring those shifts in the right way, or has a common yardstick been quietly hiding some of the most important warning signs?
How scientists usually track DNA tagging
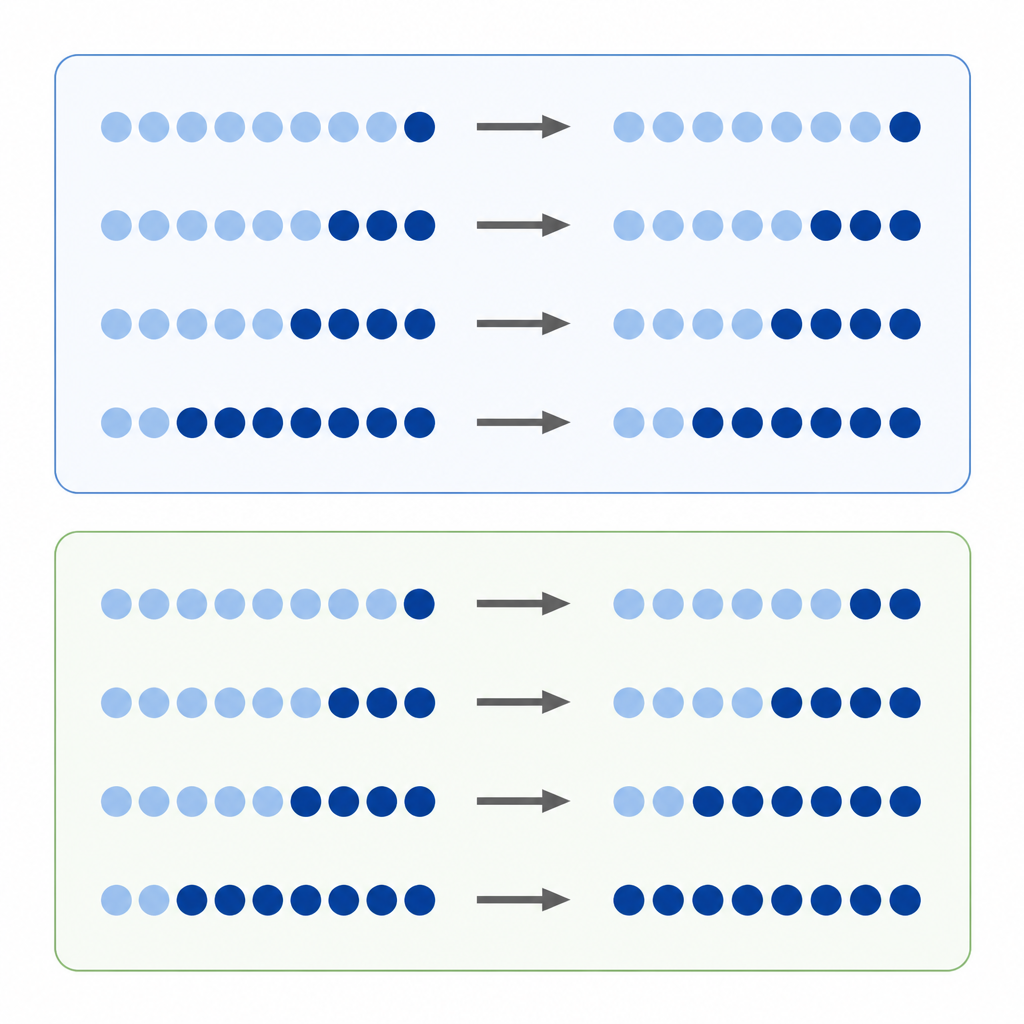
DNA methylation is one of the best-known chemical tags on our genes. Researchers often compare how much methylation a DNA site has in a tumor versus normal tissue, treating the straight difference in levels as a measure of how serious the change is. This “absolute difference” approach seems natural, but methylation levels are locked between a minimum and maximum value, and each site tends to sit at a preferred starting level. That means a site that begins highly tagged has more room to lose tags than a site that starts low, which can skew which changes look big or small.
A new way to think about change
The authors propose that many methylation shifts behave more like percentage changes than simple gains or losses. Instead of asking “how many points did this site move,” they ask “how large was the change compared with where it started?” They call this a relative change. If every site tends to gain or lose methylation at roughly the same percentage rate, then focusing only on absolute differences will favor some sites and overlook others, creating a hidden bias in how we read the cancer epigenome.
Testing the idea in cells and tumors
To probe this idea, the team analyzed data from cell cultures treated with a drug that strips methylation and from thousands of tumor samples representing many cancer types. In treated cells, sites that began with high methylation showed the biggest absolute drops, just as expected. But when the researchers looked at relative changes, they saw that sites across the whole methylation range lost a similar fraction of their original tags. In large cancer datasets, the same pattern appeared: while absolute changes still depended strongly on starting levels, relative changes lined up to reveal similar percentage shifts across different sites and tumor types. Computer simulations helped show that these patterns were unlikely to come from random noise.
Finding clearer cancer signals
The authors then asked which measure does a better job of highlighting biologically meaningful changes. They compared how well absolute and relative changes detected known methylation “signatures” linked to factors like age, smoking, and a special pattern in colorectal cancer. Relative changes were more sensitive at the extremes, such as sites that were nearly always methylated or almost never methylated in healthy tissue. Using relative shifts also uncovered genes involved in cell adhesion, metabolism, signaling, and immune activity, all processes closely tied to how tumors grow and spread. In contrast, relying on absolute differences often pointed toward genes involved in brain-related pathways, which are harder to connect directly to cancer behavior.
Why the new view matters
By treating methylation shifts as changes relative to where each site begins, the study offers a less biased lens on the cancer genome. This perspective captures important signals in regions that standard methods tend to miss, especially where loss of methylation may destabilize chromosomes or awaken silent DNA elements. The work suggests that a large part of what we thought were the biggest methylation changes in cancer may reflect our measuring stick, not biology itself.
What this means for future research
For non-specialists, the takeaway is that how we measure change can dramatically alter the stories we tell about disease. This paper argues that DNA methylation in cancer usually shifts by a fairly consistent percentage across much of the genome, and that focusing on relative change helps reveal pathways more clearly tied to tumor growth and spread. Adopting this new approach could sharpen future efforts to use methylation patterns to understand cancer risk, track tumor evolution, and perhaps guide diagnosis and treatment, without changing the underlying data at all.
Citation: Downs, B.M., Hu, J., Park, J.S. et al. An unbiased approach to measure aberrant DNA methylation alterations. Nat Commun 17, 4522 (2026). https://doi.org/10.1038/s41467-026-71089-5
Keywords: DNA methylation, cancer epigenetics, epigenetic biomarkers, genome regulation, methylation analysis