Clear Sky Science · pl
Bezstronne podejście do mierzenia zaburzonych zmian metylacji DNA
Dlaczego drobne chemiczne znaczniki na DNA mają znaczenie
Nasze komórki noszą miliony drobnych chemicznych znaczników na DNA, które pomagają kontrolować, które geny są włączone, a które wyłączone. W raku i innych chorobach te markery mogą przesuwać się w szkodliwy sposób. W tym badaniu zadano proste, ale istotne pytanie: czy mierzymy te przesunięcia właściwie, czy też powszechnie stosowana miara cicho ukrywa niektóre z najważniejszych sygnałów ostrzegawczych?
Jak naukowcy zwykle śledzą znakowanie DNA
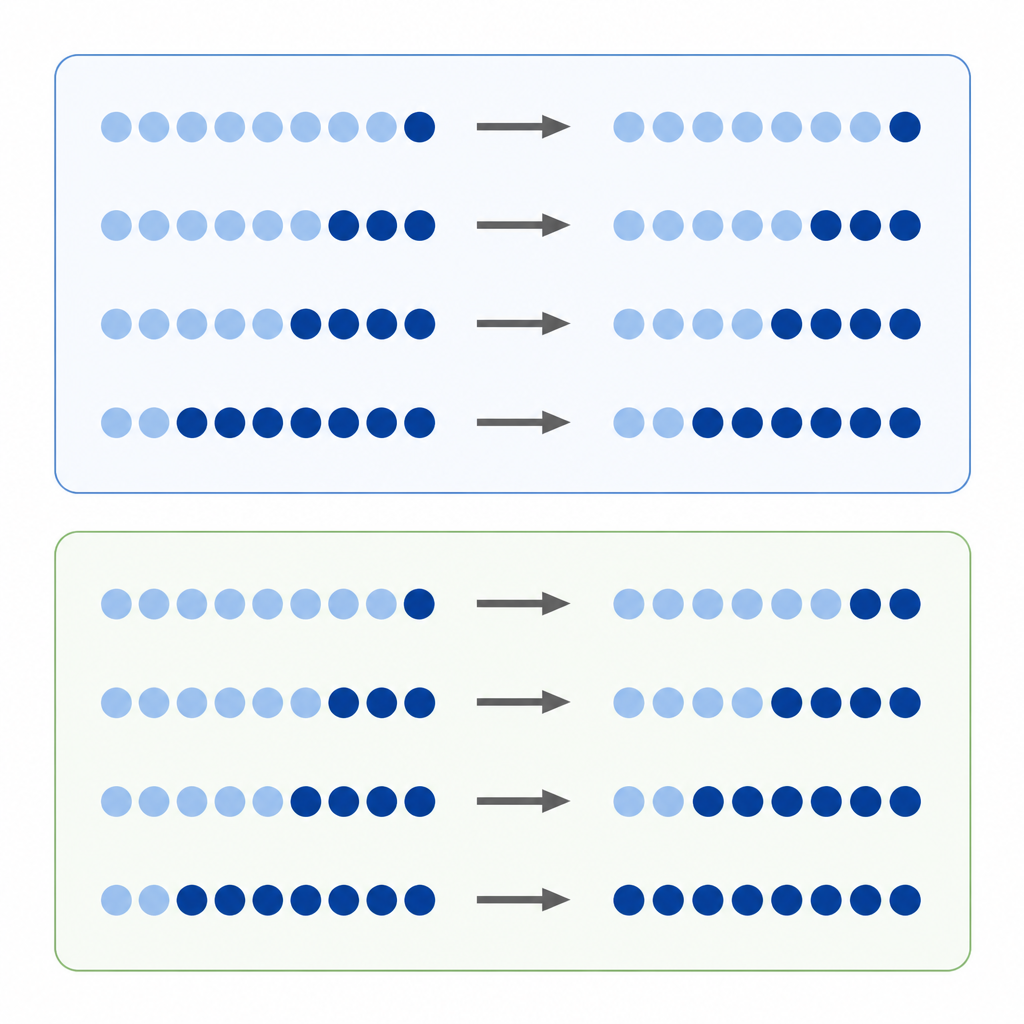
Metylacja DNA jest jednym z najlepiej znanych chemicznych znaczników na naszych genach. Badacze często porównują, ile metylacji ma dane miejsce w nowotworze w porównaniu z tkanką normalną, traktując bezwzględną różnicę poziomów jako miarę nasilenia zmiany. To podejście „różnicy bezwzględnej” wydaje się naturalne, ale poziomy metylacji są ograniczone między wartością minimalną a maksymalną, a każde miejsce ma tendencję do zajmowania preferowanego poziomu wyjściowego. Oznacza to, że miejsce zaczynające z wysoką metylacją ma więcej przestrzeni, aby utracić znaczniki niż miejsce zaczynające nisko, co może zniekształcać to, które zmiany wydają się duże lub małe.
Nowy sposób myślenia o zmianie
Autorzy proponują, że wiele przesunięć metylacji zachowuje się bardziej jak zmiany procentowe niż proste zyski czy straty. Zamiast pytać „o ile punktów przesunęło się to miejsce”, pytają „jak duża była zmiana w porównaniu z poziomem wyjściowym?”. Nazywają to zmianą względną. Jeśli każde miejsce ma tendencję do zyskiwania lub tracenia metylacji w przybliżeniu w tym samym procencie, to skupienie się wyłącznie na różnicach bezwzględnych będzie faworyzować niektóre miejsca i pomijać inne, tworząc ukrytą stronniczość w odczycie epigenomu nowotworowego.
Testowanie pomysłu w komórkach i guzach
Aby sprawdzić ten pomysł, zespół przeanalizował dane z hodowli komórkowych traktowanych lekiem usuwającym metylację oraz z tysięcy próbek guzów reprezentujących wiele typów nowotworów. W komórkach poddanych leczeniu miejsca, które zaczynały z wysoką metylacją, wykazywały największe bezwzględne spadki, zgodnie z oczekiwaniami. Jednak gdy badacze spojrzeli na zmiany względne, zobaczyli, że miejsca w całym zakresie metylacji traciły podobny ułamek swoich oryginalnych znaczników. W dużych zbiorach danych nowotworowych pojawił się ten sam wzorzec: podczas gdy zmiany bezwzględne nadal silnie zależały od poziomów wyjściowych, zmiany względne układały się tak, by ujawniać podobne procentowe przesunięcia w różnych miejscach i typach nowotworów. Symulacje komputerowe pomogły wykazać, że te wzorce były mało prawdopodobne do zaistnienia w wyniku losowego szumu.
Odnajdywanie wyraźniejszych sygnałów nowotworowych
Następnie autorzy sprawdzili, która miara lepiej podkreśla biologicznie istotne zmiany. Porównali, jak dobrze zmiany bezwzględne i względne wykrywają znane „sygnatury” metylacji związane z czynnikami takimi jak wiek, palenie czy specyficzny wzór w raku jelita grubego. Zmiany względne były bardziej czułe na skrajach, na przykład w miejscach, które w zdrowej tkance były niemal zawsze zmetylowane lub prawie nigdy zmetylowane. Użycie przesunięć względnych ujawniło również geny związane z adhezją komórkową, metabolizmem, przekazywaniem sygnałów i aktywnością układu odpornościowego — procesy ściśle powiązane z rozrostem i przerzutami guzów. W przeciwieństwie do tego, poleganie na różnicach bezwzględnych często wskazywało na geny związane ze szlakami mózgowymi, które trudniej bezpośrednio powiązać z zachowaniem nowotworu.
Dlaczego nowe spojrzenie ma znaczenie
Traktując przesunięcia metylacji jako zmiany względne względem poziomu wyjściowego każdego miejsca, badanie oferuje mniej uprzedzone spojrzenie na genom nowotworu. Ta perspektywa wychwytuje ważne sygnały w regionach, które standardowe metody mają tendencję przegapiać, szczególnie tam, gdzie utrata metylacji może destabilizować chromosomy lub uaktywniać milczące elementy DNA. Praca sugeruje, że duża część tego, co uznawaliśmy za największe zmiany metylacji w raku, może odzwierciedlać naszą miarę, a nie samą biologię.
Co to oznacza dla przyszłych badań
Dla osób niezwiązanych ze specjalistycznym polem wniosek jest taki, że sposób mierzenia zmian może drastycznie zmienić historie, które opowiadamy o chorobie. Artykuł argumentuje, że metylacja DNA w raku zwykle zmienia się o stosunkowo stały procent w dużej części genomu i że skupienie się na zmianie względnej pomaga wyraźniej ujawnić szlaki powiązane z wzrostem i rozprzestrzenianiem się nowotworów. Przyjęcie tego nowego podejścia może wyostrzyć przyszłe wysiłki wykorzystania wzorców metylacji do rozumienia ryzyka raka, śledzenia ewolucji guza i być może wspomagania diagnozy oraz terapii, bez zmiany samych danych.
Cytowanie: Downs, B.M., Hu, J., Park, J.S. et al. An unbiased approach to measure aberrant DNA methylation alterations. Nat Commun 17, 4522 (2026). https://doi.org/10.1038/s41467-026-71089-5
Słowa kluczowe: metylacja DNA, epigenetyka nowotworów, biomarkery epigenetyczne, regulacja genomu, analiza metylacji