Clear Sky Science · it
Un approccio imparziale per misurare le alterazioni aberranti della metilazione del DNA
Perché contano i piccoli marcatori chimici sul DNA
Le nostre cellule portano milioni di piccoli marcatori chimici sul loro DNA che aiutano a controllare quali geni sono attivi o inattivi. Nel cancro e in altre malattie, questi marcatori possono spostarsi in modi dannosi. Questo studio pone una domanda semplice ma importante: stiamo misurando questi spostamenti nel modo giusto, o una comune misura di riferimento ha silenziosamente nascosto alcuni dei segnali d’allarme più rilevanti?
Come gli scienziati seguono di solito l’etichettatura del DNA
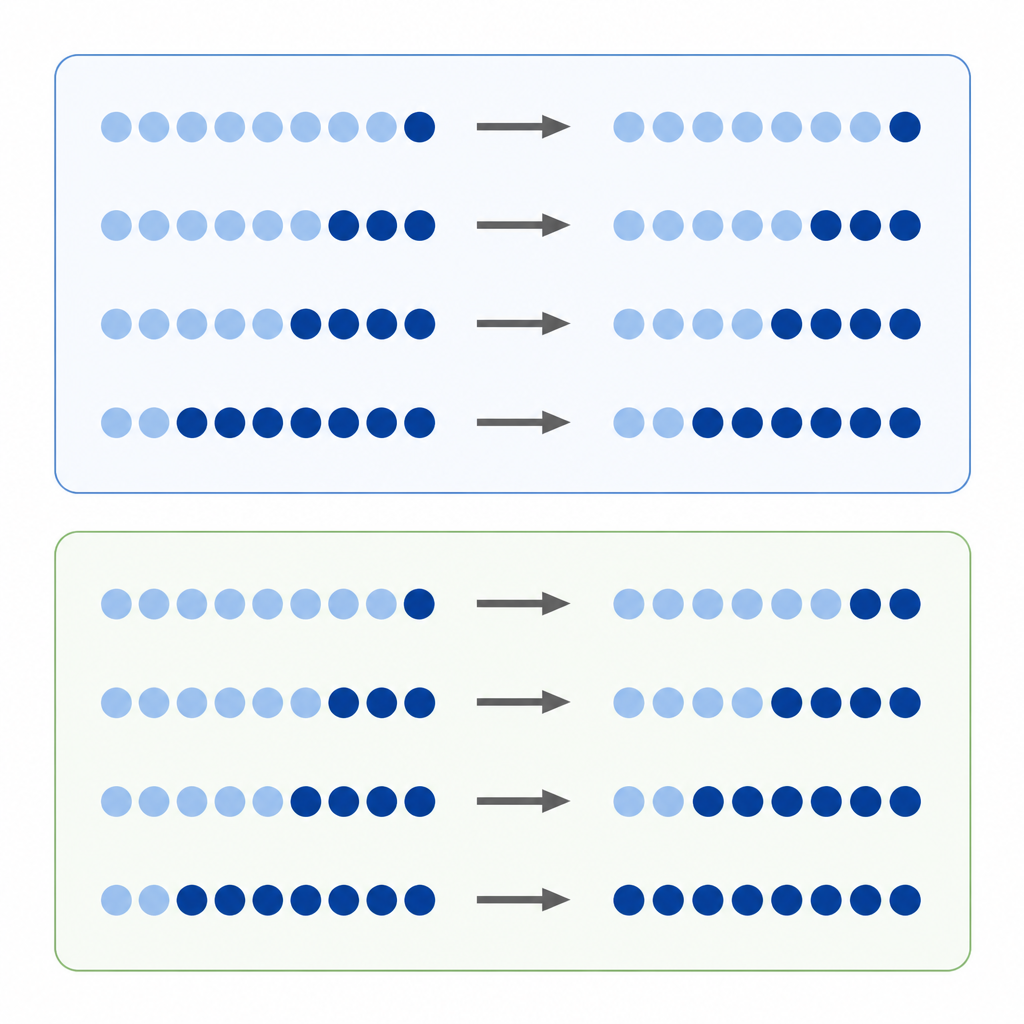
La metilazione del DNA è uno dei marcatori chimici più noti sui nostri geni. I ricercatori spesso confrontano quanto è metilato un sito del DNA in un tumore rispetto al tessuto normale, trattando la differenza diretta nei livelli come una misura della gravità del cambiamento. Questo approccio della “differenza assoluta” sembra naturale, ma i livelli di metilazione sono vincolati tra un minimo e un massimo e ogni sito tende a trovarsi a un livello iniziale preferito. Ciò significa che un sito che inizia altamente metilato ha più margine per perdere marcatori rispetto a un sito che parte basso, il che può distorcere quali cambiamenti appaiono grandi o piccoli.
Un nuovo modo di pensare al cambiamento
Gli autori propongono che molti spostamenti di metilazione si comportino più come cambiamenti percentuali che come semplici guadagni o perdite. Invece di chiedersi “di quanti punti si è spostato questo sito”, chiedono “quanto è grande il cambiamento rispetto al punto di partenza?”. Lo chiamano cambiamento relativo. Se ogni sito tende a guadagnare o perdere metilazione a un tasso percentuale approssimativamente simile, allora concentrarsi solo sulle differenze assolute favorirà alcuni siti e ne trascurerà altri, creando un bias nascosto nel modo in cui interpretiamo l’epigenoma del cancro.
Testare l’idea in cellule e tumori
Per indagare l’idea, il team ha analizzato dati da colture cellulari trattate con un farmaco che rimuove la metilazione e da migliaia di campioni tumorali rappresentativi di molti tipi di cancro. Nelle cellule trattate, i siti che iniziavano con alta metilazione mostrarono i maggiori cali assoluti, come previsto. Ma quando i ricercatori guardarono i cambiamenti relativi, videro che siti in tutto l’intervallo di metilazione persero una frazione simile dei loro marcatori originali. Nei grandi set di dati tumorali, emerse lo stesso schema: mentre i cambiamenti assoluti dipendevano ancora fortemente dai livelli iniziali, i cambiamenti relativi si allineavano rivelando simili spostamenti percentuali attraverso diversi siti e tipi di tumore. Simulazioni al computer contribuirono a mostrare che questi schemi difficilmente potevano derivare dal rumore casuale.
Trovare segnali tumorali più chiari
Gli autori hanno poi chiesto quale misura fosse più efficace nel mettere in evidenza cambiamenti biologicamente significativi. Hanno confrontato quanto bene cambiamenti assoluti e relativi rilevassero note “firme” di metilazione collegate a fattori come età, fumo e un pattern particolare nel carcinoma colorettale. I cambiamenti relativi si sono mostrati più sensibili agli estremi, come i siti quasi sempre metilati o quasi mai metilati nei tessuti sani. L’uso degli spostamenti relativi ha inoltre portato alla luce geni coinvolti nell’adesione cellulare, nel metabolismo, nella segnalazione e nell’attività immunitaria, processi strettamente legati alla crescita e alla diffusione dei tumori. Al contrario, affidarsi alle differenze assolute spesso indirizzava verso geni coinvolti in vie correlate al cervello, più difficili da collegare direttamente al comportamento tumorale.
Perché la nuova prospettiva conta
Trattando gli spostamenti di metilazione come cambiamenti relativi al livello iniziale di ciascun sito, lo studio offre una lente meno distorta sul genoma tumorale. Questa prospettiva cattura segnali importanti in regioni che i metodi standard tendono a perdere, specialmente dove la perdita di metilazione può destabilizzare i cromosomi o risvegliare elementi di DNA silenti. Il lavoro suggerisce che gran parte di ciò che consideravamo i maggiori cambiamenti di metilazione nel cancro potrebbe riflettere il nostro metro di misura, non la biologia stessa.
Cosa significa per le ricerche future
Per i non specialisti, la conclusione è che il modo in cui misuriamo il cambiamento può alterare radicalmente le storie che raccontiamo sulla malattia. Questo articolo sostiene che la metilazione del DNA nel cancro di solito cambia con una percentuale abbastanza costante in gran parte del genoma e che concentrarsi sul cambiamento relativo aiuta a svelare vie più chiaramente legate alla crescita e alla diffusione tumorale. Adottare questo nuovo approccio potrebbe affinare i futuri sforzi per usare i modelli di metilazione per comprendere il rischio di cancro, monitorare l’evoluzione del tumore e forse guidare diagnosi e terapie, senza modificare affatto i dati sottostanti.
Citazione: Downs, B.M., Hu, J., Park, J.S. et al. An unbiased approach to measure aberrant DNA methylation alterations. Nat Commun 17, 4522 (2026). https://doi.org/10.1038/s41467-026-71089-5
Parole chiave: Metilazione del DNA, Epigenetica del cancro, Biomarcatori epigenetici, Regolazione del genoma, Analisi della metilazione