Clear Sky Science · de
Ein unvoreingenommener Ansatz zur Messung abnormer DNA-Methylierungsveränderungen
Warum winzige chemische Marker auf der DNA wichtig sind
Unsere Zellen tragen Millionen kleiner chemischer Marker auf ihrer DNA, die mitbestimmen, welche Gene an- oder ausgeschaltet sind. Bei Krebs und anderen Erkrankungen können sich diese Marker in schädlicher Weise verschieben. Die Studie stellt eine einfache, aber wichtige Frage: Messen wir diese Verschiebungen richtig, oder hat ein gängiges Messmaß einige der wichtigsten Warnsignale unbemerkt verdeckt?
Wie Wissenschaftlerinnen und Wissenschaftler üblicherweise DNA‑Markierung verfolgen
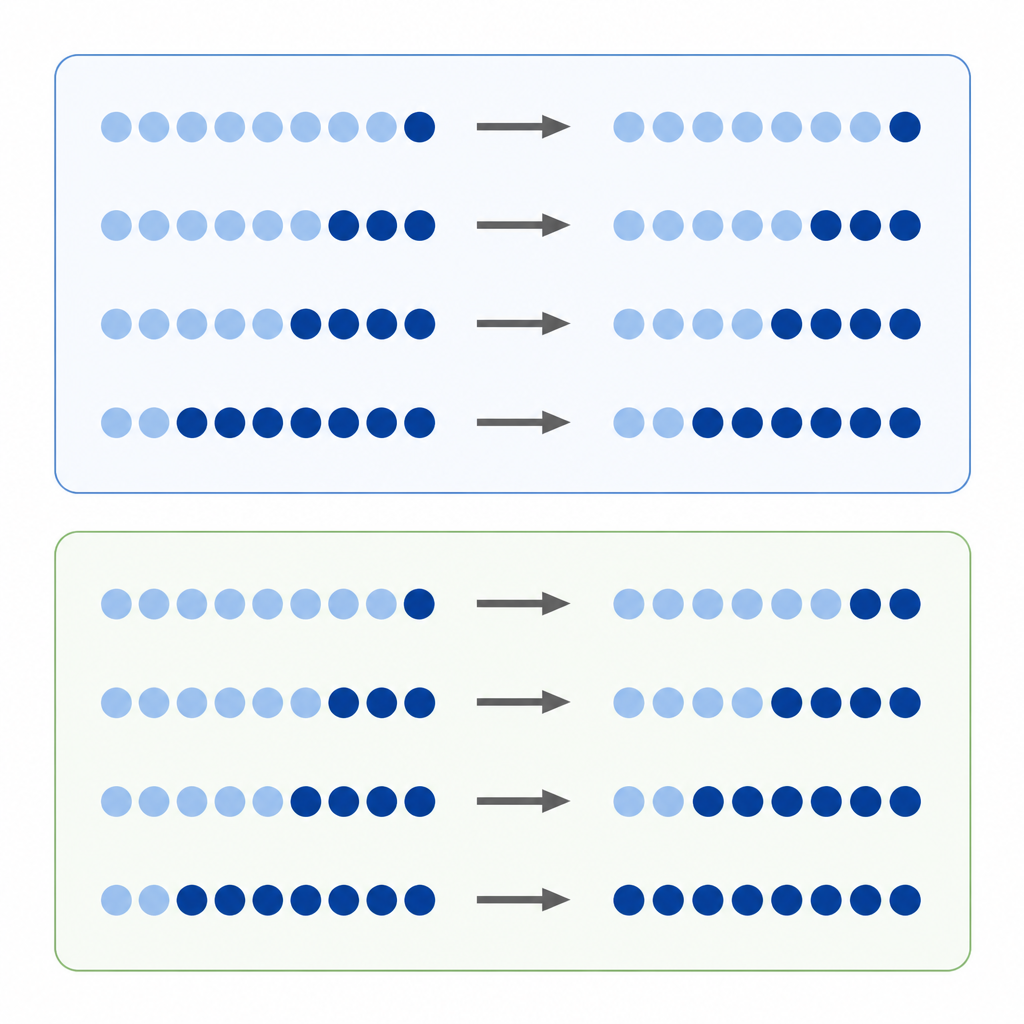
DNA‑Methylierung ist einer der bekanntesten chemischen Marker auf unseren Genen. Forschende vergleichen oft, wie stark ein DNA‑Ort in einem Tumor im Vergleich zu normalem Gewebe methyliert ist und werten die direkte Differenz als Maß für die Bedeutung der Veränderung. Dieser Ansatz der „absoluten Differenz“ wirkt naheliegend, doch Methylierungsgrade sind zwischen einem Minimum und Maximum begrenzt und jeder Ort hat typischerweise ein bevorzugtes Anfangsniveau. Das bedeutet: Ein stark methyliertes Gebiet hat mehr Spielraum, Marker zu verlieren, als ein Bereich mit niedrigem Ausgangsniveau—was verzerrt, welche Veränderungen groß oder klein erscheinen.
Eine neue Denkweise für Veränderungen
Die Autorinnen und Autoren schlagen vor, dass viele Methylierungsverschiebungen sich eher wie prozentuale Änderungen verhalten als wie einfache Zuwächse oder Verluste. Statt zu fragen „um wie viele Punkte hat sich dieser Ort verschoben“, fragen sie „wie groß war die Änderung im Verhältnis zum Ausgangswert?“ Sie nennen dies eine relative Änderung. Wenn jeder Ort dazu neigt, Methylierung in etwa gleicher prozentualer Höhe zu gewinnen oder zu verlieren, dann begünstigt die Fokussierung allein auf absolute Differenzen bestimmte Orte und übersieht andere—es entsteht eine versteckte Verzerrung in der Interpretation des Krebs‑Epigenoms.
Die Idee in Zellen und Tumoren testen
Um die Idee zu prüfen, analysierte das Team Daten aus Zellkulturen, die mit einem Wirkstoff behandelt wurden, der Methylierung entfernt, sowie Tausende von Tumorproben aus vielen Krebsarten. In behandelten Zellen zeigten Orte mit hohem Ausgangs‑Methylierungsniveau die größten absoluten Abfälle, wie zu erwarten war. Betrachtete man jedoch die relativen Änderungen, so verlor eine breite Palette von Orten über das gesamte Methylierungs‑Spektrum hinweg einen ähnlichen Bruchteil ihrer ursprünglichen Marker. In großen Krebsdatensätzen zeigte sich dasselbe Muster: Während absolute Änderungen weiterhin stark vom Ausgangsniveau abhingen, deckten relative Änderungen vergleichbare prozentuale Verschiebungen über verschiedene Orte und Tumortypen hinweg auf. Computersimulationen unterstützten, dass diese Muster kaum zufällig zu erwarten sind.
Klarere Krebs‑Signale finden
Die Autorinnen und Autoren fragten anschließend, welche Messgröße biologisch sinnvollere Veränderungen hervorhebt. Sie verglichen, wie gut absolute und relative Änderungen bekannte Methylierungs‑„Signaturen“ erkennen, die mit Faktoren wie Alter, Rauchen und einem speziellen Muster beim kolorektalen Krebs verknüpft sind. Relative Änderungen waren empfindlicher an den Extremen, etwa für Orte, die im gesunden Gewebe fast immer oder nahezu nie methyliert sind. Die Betrachtung relativer Verschiebungen förderte außerdem Gene zutage, die an Zelladhäsion, Stoffwechsel, Signalübertragung und Immunaktivität beteiligt sind—Prozesse, die eng mit Tumorwachstum und Metastasierung verbunden sind. Im Gegensatz dazu zeigten absolute Differenzen häufiger Signale in gehirnbezogenen Wegen, die sich schwerer direkt mit Krebsverhalten verknüpfen lassen.
Warum die neue Sichtweise wichtig ist
Indem Methylierungsverschiebungen relativ zu ihrem Ausgangswert betrachtet werden, bietet die Studie eine weniger verzerrte Perspektive auf das Krebs‑Genom. Diese Sicht fängt wichtige Signale in Regionen ein, die Standardmethoden tendenziell übersehen, insbesondere dort, wo Methylierungsverlust Chromosomen destabilisieren oder stille DNA‑Elemente aktivieren könnte. Die Arbeit legt nahe, dass ein großer Teil dessen, was wir für die stärksten Methylierungsänderungen bei Krebs hielten, vielleicht unser Messinstrument widerspiegelt und nicht die Biologie selbst.
Was das für künftige Forschung heißt
Für Nicht‑Spezialisten lautet die Botschaft: Wie wir Änderung messen, kann die Geschichten, die wir über Krankheiten erzählen, stark verändern. Dieses Paper argumentiert, dass sich die DNA‑Methylierung bei Krebs über große Teile des Genoms meistens um einen recht konstanten Prozentsatz verschiebt und dass die Fokussierung auf relative Änderungen Wege deutlicher sichtbar macht, die mit Tumorwachstum und -ausbreitung verbunden sind. Die Übernahme dieses neuen Ansatzes könnte künftige Bemühungen verbessern, Methylierungsmuster zu nutzen, um Krebsrisiko zu verstehen, die Tumorentwicklung zu verfolgen und möglicherweise Diagnostik und Therapie zu unterstützen—ohne die zugrundeliegenden Daten zu verändern.
Zitation: Downs, B.M., Hu, J., Park, J.S. et al. An unbiased approach to measure aberrant DNA methylation alterations. Nat Commun 17, 4522 (2026). https://doi.org/10.1038/s41467-026-71089-5
Schlüsselwörter: DNA methylation, cancer epigenetics, epigenetic biomarkers, genome regulation, methylation analysis