Clear Sky Science · pl
Warianty bialleliczne w RNU2-2 powodują najczęściej występujące znane recesywne zaburzenie rozwoju neurologicznego
Ukryte wskazówki w DNA rodziny
Niektóre z najpoważniejszych dziecięcych schorzeń mózgu przez długi czas pozostawały bez wyjaśnienia, pozostawiając rodziny bez jasnych odpowiedzi i wskazówek. To badanie odkrywa jedno z najczęstszych dziedzicznych źródeł takich zaburzeń, wskazując przyczynę nie w genie kodującym białko, lecz w małej cząsteczce RNA, która pomaga komórkom przetwarzać informacje genetyczne. Zrozumienie tego nowego zaburzenia nie tylko daje długo oczekiwane diagnozy wielu rodzinom, lecz także otwiera praktyczne możliwości testów nosicielstwa, planowania rodziny oraz wcześniejszej opieki nad dotkniętymi dziećmi. 
Maleńki RNA o wielkiej roli
Nasze komórki muszą poprawiać surowe informacje genetyczne zanim zostaną użyte do tworzenia białek. Ten proces, zwany składaniem (splicingiem), wykonuje potężna maszyna znana jako spliceosom. Jednym z jej kluczowych składników jest U2 mały RNA jądrowy, krótka cząsteczka RNA, która pomaga rozpoznać miejsca, gdzie fragmenty kodu genetycznego powinny być wycięte i połączone. Gen RNU2-2 koduje jedną wersję tego RNA U2. Do niedawna zmiany w tym i powiązanych genach RNA były znane jako przyczyna zaburzeń mózgu o dominującym charakterze — gdzie jedna wadliwa kopia wystarcza do wystąpienia choroby. Nowe badanie ujawnia, że gdy obie kopie RNU2-2 są uszkodzone, pojawia się inna, recesywna forma zaburzenia rozwojowego układu nerwowego, i że jest ona zaskakująco powszechna.
Odkrycie powszechnie występującego dziedzicznego zaburzenia mózgu
Naukowcy przeanalizowali dane genetyczne dziesiątek tysięcy osób z rzadkimi schorzeniami zapisanych do brytyjskiego projektu 100 000 Genomów oraz do programów genomowych National Health Service. Korzystając z narzędzia statystycznego zaprojektowanego do wykrywania rzadkich, chorobotwórczych wariantów, porównali geny niekodujące białek u ponad 14 000 osób z problemami rozwojowymi układu nerwowego z ponad 50 000 osób bez takich rozpoznań. Wyraźnie wyróżniły się tylko dwa geny, RNU4-2 i RNU2-2, ale kiedy skoncentrowali się na zmianach działających w sposób recesywny — gdzie osoba musi odziedziczyć jedną zmienioną kopię od każdego z rodziców — RNU2-2 dał przytłaczające dowody. Zidentyfikowali 18 rodzin o wysokim stopniu pewności, w których chore dzieci miały dwie szkodliwe zmiany w RNU2-2 na przeciwległych kopiach genu, a także dodatkowe rodziny kandydujące oraz dziewięć kolejnych przypadków z niezależnych kohort w Stanach Zjednoczonych, we Włoszech i w Holandii. 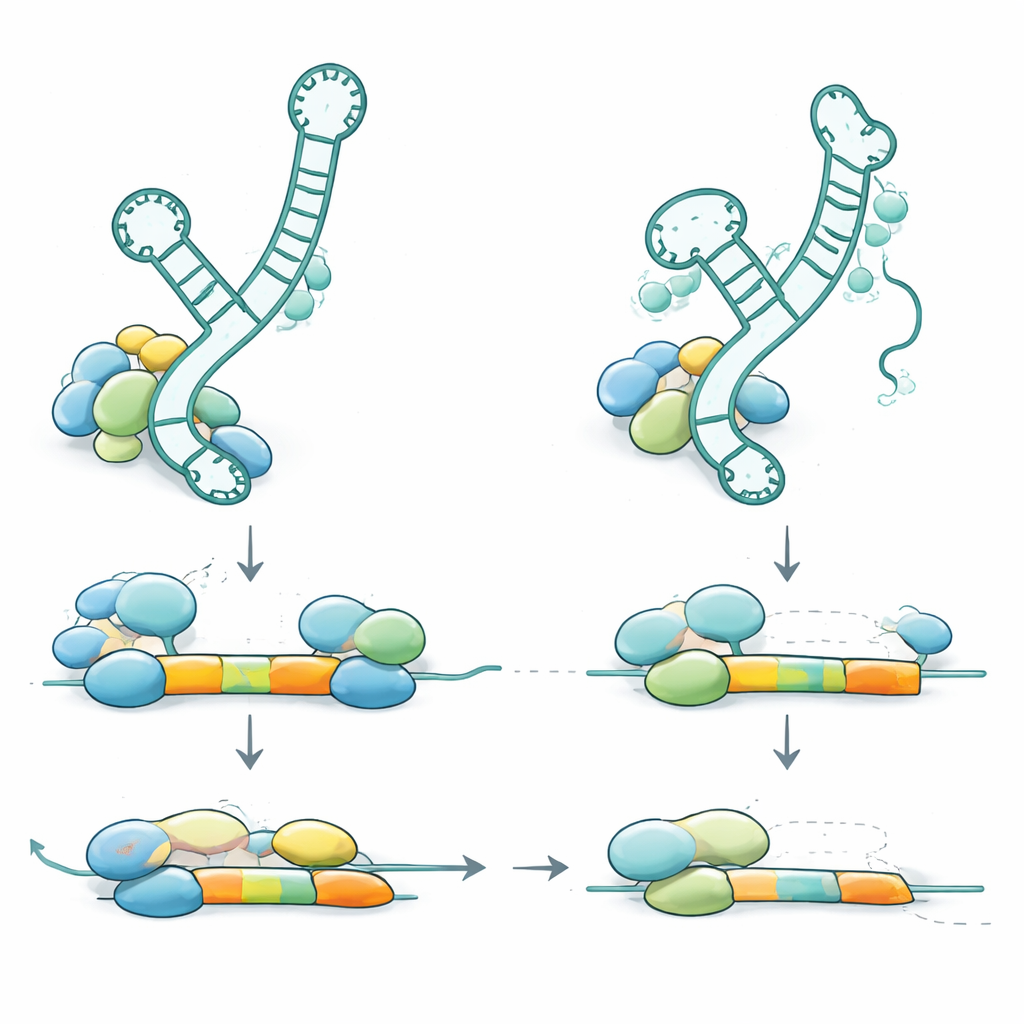
Jak choroba wygląda w praktyce
Dzieci z recesywnym zespołem RNU2-2 zwykle trafiają pod opiekę medyczną w niemowlęctwie lub wczesnym dzieciństwie. Większość ma opóźnienia w osiąganiu kamieni milowych rozwoju, takich jak siedzenie, chodzenie czy mówienie, i wiele z nich ma umiarkowaną do ciężkiej niepełnosprawność intelektualną. Napady padaczkowe są bardzo częste i mogą rozpoczynać się wcześnie; u niektórych dzieci rozwijają się w trudne do leczenia zespoły padaczkowe. Napięcie mięśniowe i ruchy są często zaburzone, od niskiego napięcia w niemowlęctwie po sztywność, nieprawidłowe postawy lub mimowolne ruchy w późniejszym okresie. Obrazowanie mózgu może początkowo wyglądać normalnie, lecz z czasem może ujawnić ubytek tkanki mózgowej lub zmiany w istocie białej łączącej różne regiony. Niektórzy pacjenci mają łagodniejsze objawy i pozostają stosunkowo stabilni, podczas gdy inni rozwijają poważne powikłania, w tym problemy z oddychaniem, karmieniem i — w rzadkich przypadkach — wczesną śmierć.
W jaki sposób zmiany w RNU2-2 zakłócają komórkowe poprawki
Aby zrozumieć, jak te zmiany genetyczne powodują chorobę, zespół zbadał, gdzie występują warianty w RNA U2-2 i jak wpływają na jego strukturę oraz zachowanie. Wiele wariantów recesywnych prawdopodobnie osłabia struktury pętli‑łodyg (stem‑loop) — maleńkie włoski w RNA, które pomagają mu wiązać białka partnerów i inne RNA. Inne leżą bezpośrednio w regionie rozpoznającym miejsca splicingowe lub w miejscu dokowania pierścienia białek pomocniczych. Gdy badacze zbadali próbki krwi, stwierdzili, że wadliwe kopie RNU2-2 były wyrażane na poziomie poniżej 10% normalnego u osób dotkniętych, co wskazuje, że zmienione RNA jest niestabilne i w dużej mierze niszczone. Organizm częściowo rekompensuje to zwiększając ekspresję powiązanego genu U2 (RNU2-1), więc całkowity poziom U2 pozostaje w przybliżeniu normalny, lecz ta kompensacja nie wystarcza, by zapobiec chorobie. U zdrowych nosicieli z jedną wadliwą kopią uszkodzone RNA również jest silnie zredukowane, jednak pozostająca prawidłowa kopia zwiększa swoją produkcję, utrzymując funkcję powyżej progu występowania objawów.
Dlaczego to odkrycie ma znaczenie dla rodzin
Ponieważ ten zespół ma dziedziczenie recesywne, ma tendencję do występowania wśród rodzeństwa, gdy oboje rodzice są zdrowymi nosicielami. W brytyjskim projekcie genomowym zespół RNU2-2 recesywny odpowiada za około jedną na dziesięć rodzin z rozpoznaną recesywną przyczyną zaburzeń rozwoju neurologicznego, co czyni go najczęstszą pojedynczą przyczyną w tej kategorii i niemal tak powszechnym jak wcześniej opisane dominujące zaburzenie spowodowane przez inny gen RNA spliceosomalnego. Co kluczowe, autorzy pokazują, że prosty test RNA z krwi może pomóc odróżnić naprawdę szkodliwe warianty od łagodnych, mierząc, ile RNU2-2 zostało utracone oraz jak silnie zwiększa się kompensacyjny U2-1. Dla rodzin oznacza to jaśniejsze diagnozy, lepsze oszacowania ryzyka nawrotu oraz możliwość poradnictwa genetycznego przed poczęciem lub w czasie ciąży — przekształcając niejasny kawałek biologii RNA w praktyczne informacje do wykorzystania w decyzjach medycznych w realnym świecie.
Cytowanie: Greene, D., Mendez, R., Lees, J. et al. Biallelic variants in RNU2-2 cause the most prevalent known recessive neurodevelopmental disorder. Nat Genet 58, 774–781 (2026). https://doi.org/10.1038/s41588-026-02539-5
Słowa kluczowe: zaburzenie rozwoju neurologicznego, dziedziczenie recesywne, spliceosom, mały RNA jądrowy, sekwencjonowanie genomu