Clear Sky Science · fr
Variants bialléliques dans RNU2-2 causent le trouble neurodéveloppemental récessif connu le plus fréquent
Indices cachés dans l’ADN familial
Certaines des affections cérébrales infantiles les plus graves sont demeurées longtemps inexpliquées, laissant les familles sans réponses ni orientations claires. Cette étude révèle l’une des causes héréditaires les plus courantes de ces troubles, en la reliant non pas à un gène codant pour une protéine mais à un minuscule fragment d’ARN qui aide les cellules à traiter les messages génétiques. Comprendre ce nouveau trouble offre non seulement des diagnostics longtemps attendus pour de nombreuses familles, mais ouvre aussi des possibilités pratiques de dépistage des porteurs, de planification familiale et de prise en charge plus précoce des enfants affectés. 
Un petit ARN aux grandes fonctions
Nos cellules doivent éditer les messages génétiques bruts avant de pouvoir les utiliser pour fabriquer des protéines. Cette édition, appelée épissage, est réalisée par une vaste machinerie connue sous le nom de spliceosome. L’un de ses composants clés est l’ARN nucléaire U2, une courte molécule d’ARN qui aide à reconnaître où les segments de code génétique doivent être coupés et rattachés. Le gène RNU2-2 code une version de cet ARN U2. Jusqu’à récemment, les altérations de ce gène et de gènes ARN apparentés étaient connues pour provoquer des troubles cérébraux de façon dominante — où une seule copie défectueuse suffit à déclencher la maladie. Le nouveau travail révèle que lorsque les deux copies de RNU2-2 sont endommagées, apparaît une forme différente, récessive, de trouble neurodéveloppemental, et qu’elle est étonnamment fréquente.
Découverte d’un trouble cérébral héréditaire fréquent
Les chercheurs ont passé au crible les données génétiques de dizaines de milliers de personnes atteintes de maladies rares inscrites au projet britannique 100 000 Genomes et aux programmes génomiques du National Health Service. À l’aide d’un outil statistique conçu pour détecter des variants rares causant des maladies, ils ont comparé les gènes non codants pour des protéines chez plus de 14 000 individus présentant des troubles du neurodéveloppement à plus de 50 000 personnes sans tels diagnostics. Seuls deux gènes se sont démarqués, RNU4-2 et RNU2-2, mais lorsqu’ils ont recherché spécifiquement des changements agissant de manière récessive — où une personne doit hériter d’une copie altérée de chaque parent — RNU2-2 a présenté des preuves accablantes. Ils ont identifié 18 familles à haute confiance dans lesquelles des enfants affectés portaient deux variants délétères de RNU2-2 sur des copies opposées du gène, ainsi que des familles candidates supplémentaires et neuf cas de plus provenant de cohortes indépendantes aux États-Unis, en Italie et aux Pays-Bas. 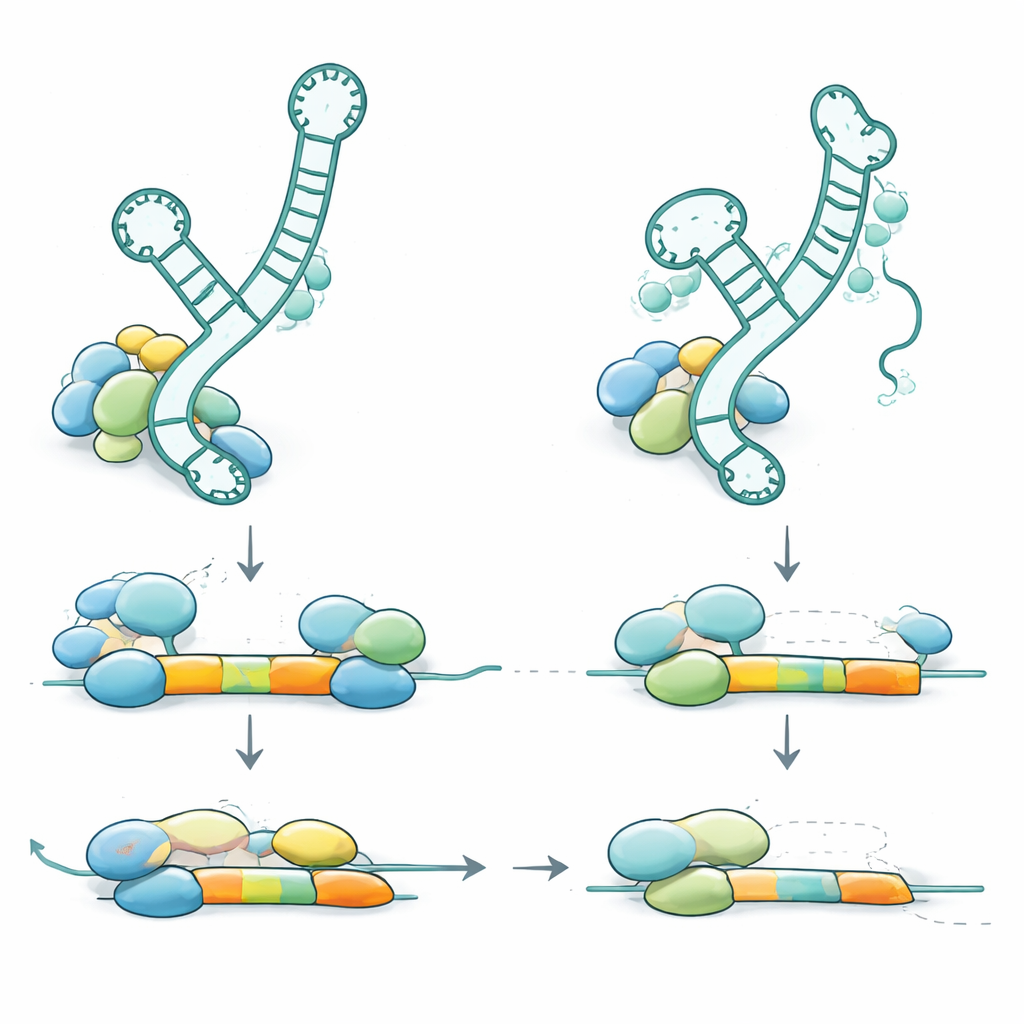
Manifestations cliniques du trouble
Les enfants atteints de ce syndrome récessif lié à RNU2-2 sont généralement pris en charge médicalement pendant l’enfance précoce. La plupart présentent des retards dans l’acquisition des étapes du développement, comme s’asseoir, marcher ou parler, et beaucoup ont une déficience intellectuelle modérée à sévère. Les crises épileptiques sont très fréquentes et peuvent débuter tôt ; chez certains enfants elles évoluent vers des syndromes épileptiques difficiles à traiter. Le tonus et le mouvement musculaire sont souvent perturbés, allant d’un faible tonus à la naissance à de la raideur, des postures anormales ou des mouvements involontaires ultérieurs. Les examens cérébraux peuvent paraître normaux au départ mais montrer par la suite une perte de tissu cérébral ou des altérations de la matière blanche qui relie les différentes régions. Certaines personnes ont une atteinte légère et restent relativement stables, tandis que d’autres développent des complications sévères, notamment des problèmes respiratoires, d’alimentation et, dans de rares cas, un décès précoce.
Comment les changements dans RNU2-2 perturbent l’édition cellulaire
Pour comprendre comment ces variations génétiques provoquent la maladie, l’équipe a examiné où se situent les variants au sein de l’ARN U2-2 et comment ils affectent sa structure et son comportement. Beaucoup des variants récessifs devraient fragiliser des structures en tige-boucle — de petites épingles à cheveux dans l’ARN qui aident à lier des protéines partenaires et d’autres ARN. D’autres se situent directement dans la région qui reconnaît les sites d’épissage ou dans le site d’arrimage pour un anneau de protéines auxiliaires. Lorsqu’ils ont étudié des échantillons de sang, ils ont constaté que les copies défectueuses de RNU2-2 étaient exprimées à moins de 10 % des niveaux normaux chez les personnes affectées, ce qui indique que l’ARN altéré est instable et largement détruit. L’organisme compense en partie en augmentant l’expression d’un gène U2 apparenté (RNU2-1), de sorte que les niveaux totaux d’U2 restent à peu près normaux, mais cette compensation ne suffit pas à prévenir la maladie. Chez les porteurs sains qui n’ont qu’une copie défectueuse, l’ARN endommagé est également fortement réduit, mais la copie normale augmente sa production, maintenant la fonction globale au‑dessus du seuil des symptômes.
Pourquoi cette découverte compte pour les familles
Parce que ce syndrome suit un héritage récessif, il tend à se reproduire chez les frères et sœurs lorsque les deux parents sont des porteurs sains. Dans le projet génome au Royaume‑Uni, le syndrome récessif RNU2-2 représente environ une famille sur dix avec un diagnostic récessif connu de trouble neurodéveloppemental, ce qui en fait la cause unique la plus fréquente dans cette catégorie et presque aussi courante qu’un trouble dominant décrit précédemment causé par un autre gène d’ARN du spliceosome. De façon cruciale, les auteurs montrent qu’un simple test d’ARN sanguin peut aider à distinguer les variants réellement nuisibles des variants bénins en mesurant combien de RNU2-2 est perdu et dans quelle mesure le gène de secours U2-1 est surexprimé. Pour les familles, cela signifie des diagnostics plus clairs, de meilleures estimations du risque de récurrence et la possibilité de conseils génétiques préconceptionnels ou prénataux — transformant un fragment obscur de biologie de l’ARN en des informations exploitables pour des décisions médicales concrètes.
Citation: Greene, D., Mendez, R., Lees, J. et al. Biallelic variants in RNU2-2 cause the most prevalent known recessive neurodevelopmental disorder. Nat Genet 58, 774–781 (2026). https://doi.org/10.1038/s41588-026-02539-5
Mots-clés: trouble neurodéveloppemental, héritage récessif, spliceosome, petit ARN nucléaire, séquençage du génome