Clear Sky Science · pl
Badanie chemii i katalizy przez uprzedzanie skośnych rozkładów za pomocą głębokiego uczenia
Dlaczego to nowe narzędzie chemiczne ma znaczenie
Chemicy chcą wiedzieć nie tylko, jak działają reakcje, lecz także które reakcje są w ogóle możliwe, gdy cząsteczki spotykają się w rzeczywistych warunkach — w rozpuszczalnikach, na powierzchniach katalizatorów, w temperaturach roboczych. Tradycyjne mapowanie wszystkich sposobów, w jakie atomy mogą się przemieszczać, wymagało mozolnych przypuszczeń i ciężkich obliczeń. W artykule przedstawiono nowe podejście komputerowe, nazwane Loxodynamics, które wykorzystuje idee ze statystyki i głębokiego uczenia do automatycznego kierowania symulacjami molekularnymi w stronę najbardziej prawdopodobnych ścieżek reakcji, bez potrzeby wcześniejszej intuicji człowieka dotyczącej powstających produktów.
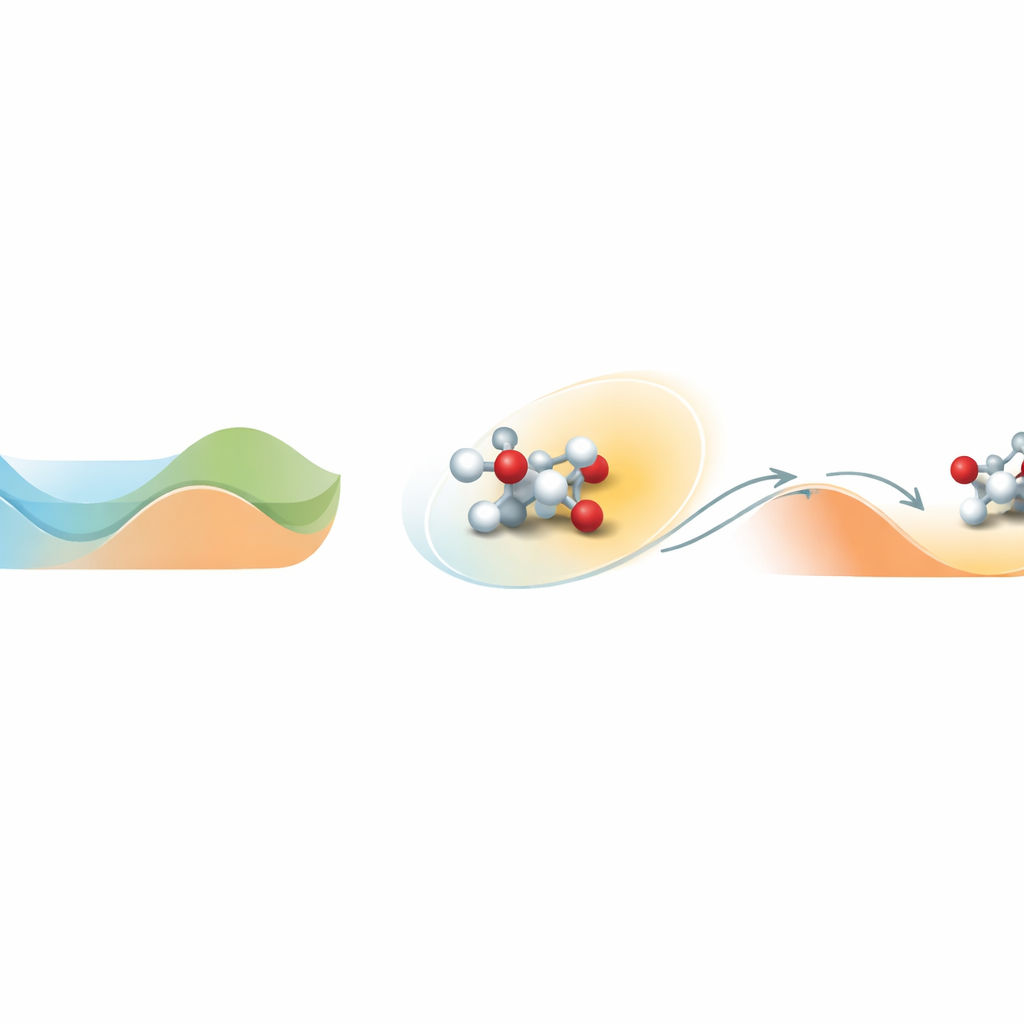
Znajdowanie wyjścia z energetycznej doliny
Istota problemu polega na tym, jak cząsteczki poruszają się po złożonym pejzażu energetycznym pełnym dolin i wzniesień. Dolina oznacza stan stabilny; wejście na wzniesienie odpowiada przekroczeniu bariery reakcyjnej. Standardowe symulacje molekularne w realistycznych temperaturach często utkną w jednej dolinie i nigdy nie zobaczą rzadkich zdarzeń przekroczenia bariery, które definiują chemię. Loxodynamics radzi sobie z tym, obserwując, jak układ wędruje w obrębie doliny, i zadając proste pytanie statystyczne: czy lokalna chmura położeń jest nieco rozciągnięta, czyli „skośna”, bardziej w jednym kierunku niż w drugim? Ta niewielka asymetria ujawnia, gdzie pobliska bariera jest najniższa i w którym kierunku układ najprawdopodobniej ucieknie.
Przekształcanie asymetrii w kierownicę
Metoda zaczyna się od krótkiej, nieobciążonej symulacji układu, podczas której zbiera się wiele migawkowych ujęć pozycji atomów i przekształca je w liczbowe deskryptory, takie jak odległości między wiązaniami. Ponieważ rzeczywiste systemy chemiczne obejmują wiele takich deskryptorów, autorzy konstruują specjalną sieć neuronową nazwaną Skewencoder. Sieć ta kompresuje dane wysokowymiarowe do pojedynczej współrzędnej, która uchwyca istotny, powolny ruch układu. Co kluczowe, Skewencoder jest trenowany nie tylko do odtwarzania danych wejściowych, lecz także do maksymalizowania skośności — miary asymetrii — rozkładu wzdłuż tej nowej współrzędnej. Gdy kierunek skośny zostanie zidentyfikowany, Loxodynamics dodaje delikatną, jednostronną harmoniczną „ścianę”, która popycha układ dalej w kierunku, gdzie ogon rozkładu jest dłuższy, skutecznie kierując go pod górę przez najniższą pobliską barierę.
Iteracyjne przeszukiwanie bez chemicznych przypuszczeń
Loxodynamics działa w cyklach „próbkuj i szukaj”. Po każdej symulacji z biasem, nowa trajektoria jest dodawana do rosnącego globalnego zestawu danych służącego do dopracowania sieci neuronowej, podczas gdy najnowszy fragment służy do aktualizacji wskazówek opartych na skośności. Ściana biasująca jest następnie przesuwana na podstawie nowego skośnego rozkładu i układ jest ponownie popychany. Ten iteracyjny proces stopniowo prowadzi układ poza jego początkową dolinę w kierunku nowych stanów metastabilnych odpowiadających pośrednikom lub produktom. Ponieważ prowadząca współrzędna i kierunek są uczone na bieżąco z danych, metoda nie wymaga od użytkownika wcześniejszego określania, które wiązania mają pękać lub powstawać ani jakich produktów oczekiwać.

Testowanie metody od prostych modeli po działające katalizatory
Aby pokazać jej siłę, autorzy najpierw stosują Loxodynamics do prostych jedno- i dwuwymiarowych pejzaży modelowych, gdzie dwie różne ścieżki łączą te same stany początkowe i końcowe. Metoda niezawodnie wybiera trasę o niższej barierze. Następnie przechodzą do rzeczywistych reakcji w fazie gazowej: klasycznej reakcji substytucji (SN2) i cykloadycji Dielsa–Aldera. Nawet przy użyciu jedynie ogólnych deskryptorów odległości Loxodynamics automatycznie znajduje poprawne ścieżki reakcji w obu kierunkach, radząc sobie z szerokimi, elastycznymi obszarami reagentów i wąskimi studniami produktów. Wreszcie podejście testowane jest w wymagających scenariuszach katalitycznych: dehydratacji etanolu i 1-butanolu wewnątrz kwaśnej struktury zeolitu. W realistycznych temperaturach roboczych metoda odkrywa zarówno znane mechanizmy „sprzężone”, jak i „krok po kroku”, identyfikuje krótkotrwałe pośredniki i rozróżnia różne produkty alkenowe — wszystko to przy użyciu prostych deskryptorów opartych na odległościach i minimalnego wkładu człowieka.
Co to oznacza dla przyszłego odkrywania reakcji
Praca ta pokazuje, że stosunkowo prosta idea — wykorzystanie skośności lokalnego ruchu do wskazania najłatwiejszej drogi ucieczki — może zostać przekształcona w praktyczne narzędzie sterujące dla symulacji molekularnych. Poprzez łączenie dynamiki z biasem ze świadomością skośności w sieci neuronowej, Loxodynamics oferuje systematyczny sposób odkrywania prawdopodobnych kanałów reakcyjnych i pośredników w warunkach skończonej temperatury, bez sztucznego podnoszenia temperatury czy wstępnego definiowania szczegółowych współrzędnych reakcji. W dłuższej perspektywie takie podejścia mogą przyspieszyć projektowanie katalizatorów, poprawić modelowane uczeniem maszynowym potencjały międzyatomowe w trudno próbkowanych rejonach przejściowych, a nawet zostać zaadaptowane do problemów niechemicznych, takich jak fałdowanie białek czy nukleacja kryształów, wszędzie tam, gdzie układy muszą znaleźć rzadkie ścieżki wyjścia z złożonych pejzaży energetycznych.
Cytowanie: Zhang, Z., Piccini, G. Exploring chemistry and catalysis by biasing skewed distributions via deep learning. Nat Commun 17, 3010 (2026). https://doi.org/10.1038/s41467-026-69586-8
Słowa kluczowe: odkrywanie reakcji, dynamika molekularna, kataliza, głębokie uczenie, powierzchnie energii swobodnej