Clear Sky Science · fr
Explorer la chimie et la catalyse en biaisant des distributions asymétriques via l’apprentissage profond
Pourquoi cet outil chimique est important
Les chimistes veulent savoir non seulement comment les réactions se déroulent, mais aussi quelles réactions sont possibles lorsque des molécules se rencontrent dans des conditions réelles — en solution, sur des surfaces catalytiques, à des températures de fonctionnement. Traditionnellement, cartographier tous les réarrangements atomiques possibles exigeait des conjectures laborieuses et des calculs lourds. Cet article présente une nouvelle approche informatique, appelée Loxodynamics, qui utilise des idées issues des statistiques et de l’apprentissage profond pour pousser automatiquement les simulations moléculaires vers les voies réactionnelles les plus probables, sans nécessiter d’intuition humaine préalable sur les produits qui se formeront.
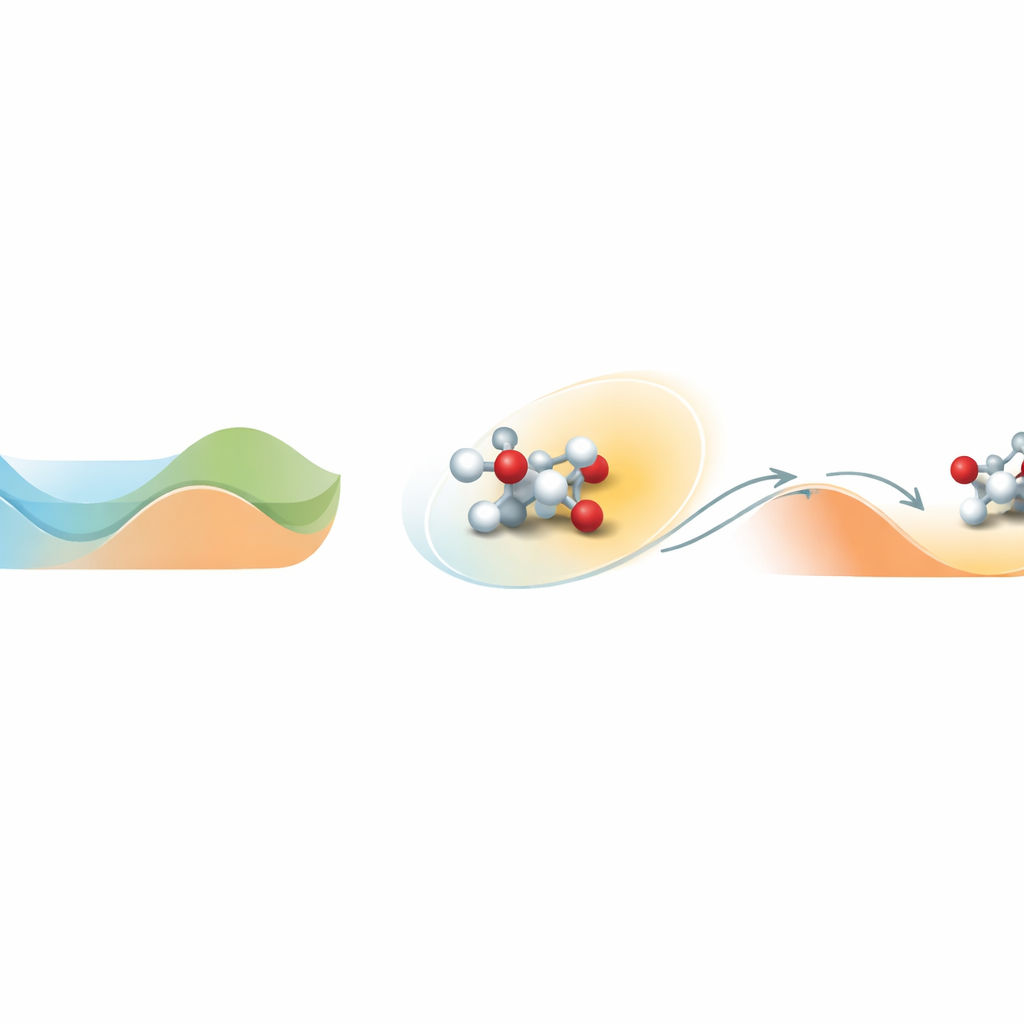
Sortir d’une vallée d’énergie
Le cœur du problème est la façon dont les molécules évoluent sur un paysage d’énergie complexe, plein de vallées et de collines. Une vallée représente un état stable ; gravir une colline correspond à franchir une barrière réactionnelle. Les simulations moléculaires standards à des températures réalistes restent souvent piégées dans une vallée et ne voient jamais les événements rares de franchissement de barrière qui définissent la chimie. Loxodynamics s’attaque à cela en observant comment un système erre à l’intérieur d’une vallée et en posant une question statistique simple : le nuage local de positions est-il légèrement étiré, ou « biaisé », plus dans une direction que dans une autre ? Cette petite asymétrie révèle où la barrière voisine est la plus basse et dans quelle direction le système est le plus susceptible de s’échapper.
Transformer l’asymétrie en volant de direction
La méthode commence par une brève simulation non biaisée du système, pendant laquelle elle collecte de nombreux instantanés des positions atomiques et les convertit en des descripteurs numériques, tels que des distances de liaison. Parce que les systèmes chimiques réels impliquent beaucoup de ces descripteurs, les auteurs construisent un réseau neuronal spécial appelé Skewencoder. Ce réseau compresse les données de haute dimension en une seule coordonnée qui capte le mouvement lent essentiel du système. De façon cruciale, le Skewencoder est entraîné non seulement à reconstruire les données d’entrée, mais aussi à maximiser l’asymétrie — une mesure de biais — de la distribution le long de cette nouvelle coordonnée. Une fois cette direction biaisée identifiée, Loxodynamics ajoute une « paroi » harmonique unilatérale douce qui pousse le système davantage dans la direction où la queue de la distribution est plus longue, le dirigeant effectivement en montée par-dessus la plus basse barrière voisine.
Recherche itérative sans conjectures chimiques
Loxodynamics progresse par cycles de « prélèvement et recherche ». Après chaque simulation biaisée, la nouvelle trajectoire est ajoutée à un jeu de données global en croissance utilisé pour affiner le réseau neuronal, tandis que le segment le plus récent sert à mettre à jour l’orientation basée sur l’asymétrie. La paroi de biais est ensuite repositionnée en fonction de la nouvelle distribution biaisée, et le système est poussé à nouveau. Ce processus itératif conduit progressivement le système hors de sa vallée initiale et vers de nouveaux états métastables correspondant à des intermédiaires ou à des produits. Parce que la coordonnée guide et la direction sont apprises en temps réel à partir des données, la méthode n’oblige pas l’utilisateur à spécifier à l’avance quelles liaisons se casseront ou se formeront, ni quels produits attendre.

Tester la méthode, des modèles simples aux catalyseurs en fonctionnement
Pour démontrer sa puissance, les auteurs appliquent d’abord Loxodynamics à des paysages modèles unidimensionnels et bidimensionnels simples, où deux chemins différents relient les mêmes états de départ et d’arrivée. La méthode identifie de manière fiable la route à barrière la plus faible. Ils passent ensuite à des réactions en phase gazeuse réelles : une réaction de substitution classique (SN2) et une cycloaddition Diels–Alder. Même alimentée uniquement avec des descripteurs de distances générales, Loxodynamics trouve automatiquement les bonnes voies réactionnelles dans les deux sens, gérant des régions réactantes larges et flexibles ainsi que des puits produits étroits. Enfin, l’approche est testée dans des scénarios catalytiques exigeants : la déshydratation de l’éthanol et du 1‑butanol à l’intérieur d’une matrice de zéolite acide. À des températures de fonctionnement réalistes, la méthode révèle des mécanismes connus « concertés » et « par étapes », identifie des intermédiaires de courte durée et distingue différents alcènes produits — tout en utilisant des descripteurs simples basés sur les distances et peu d’intervention humaine.
Ce que cela signifie pour la découverte de réactions à venir
Ce travail montre qu’une idée relativement simple — utiliser l’asymétrie du mouvement local pour indiquer la voie de sortie la plus facile — peut être transformée en un outil pratique de guidage pour les simulations moléculaires. En combinant une dynamique biaisée avec un réseau neuronal sensible à l’asymétrie, Loxodynamics offre un moyen systématique de découvrir des canaux réactionnels et des intermédiaires probables à température finie, sans élever artificiellement la température ni pré-définir des coordonnées réactionnelles détaillées. À long terme, de telles approches pourraient accélérer la conception de catalyseurs, améliorer les potentiels interatomiques appris par machine dans les régions de transition difficiles à échantillonner, et même être adaptées à des problèmes non chimiques comme le repliement des protéines ou la nucléation cristalline, partout où des systèmes doivent trouver des voies rares hors de paysages d’énergie complexes.
Citation: Zhang, Z., Piccini, G. Exploring chemistry and catalysis by biasing skewed distributions via deep learning. Nat Commun 17, 3010 (2026). https://doi.org/10.1038/s41467-026-69586-8
Mots-clés: découverte de réactions, dynamique moléculaire, catalyse, apprentissage profond, paysages d’énergie libre