Clear Sky Science · es
Explorando la química y la catálisis sesgando distribuciones asimétricas mediante aprendizaje profundo
Por qué importa esta nueva herramienta química
Los químicos quieren saber no solo cómo funcionan las reacciones, sino qué reacciones son siquiera posibles cuando las moléculas se encuentran en condiciones reales: en disolventes, sobre superficies catalíticas, a temperaturas de trabajo. Tradicionalmente, cartografiar todas las formas en que los átomos pueden reorganizarse ha exigido conjeturas meticulosas y cálculos costosos. Este artículo presenta un nuevo enfoque por ordenador, llamado Loxodynamics, que utiliza ideas de la estadística y el aprendizaje profundo para empujar automáticamente las simulaciones moleculares hacia las vías de reacción más probables, sin requerir la intuición humana previa sobre qué productos se formarán.
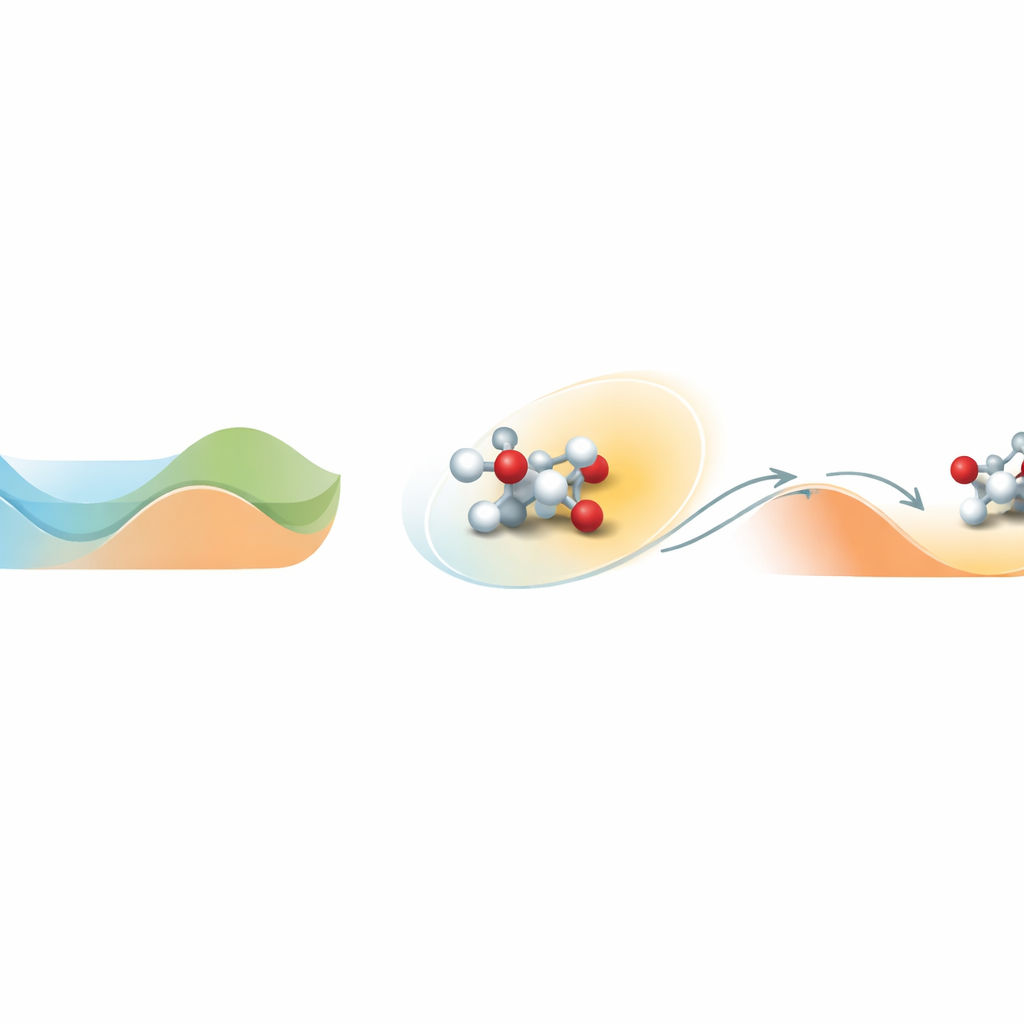
Encontrar la salida de un valle de energía
En el centro del problema está cómo se mueven las moléculas sobre un paisaje de energía complejo lleno de valles y colinas. Un valle representa un estado estable; ascender una colina corresponde a cruzar una barrera de reacción. Las simulaciones moleculares estándar a temperaturas realistas a menudo se quedan atrapadas en un valle y nunca ven los raros eventos de cruce de barrera que definen la química. Loxodynamics aborda esto observando cómo un sistema vaga dentro de un valle y formulando una simple pregunta estadística: ¿la nube local de posiciones está ligeramente estirada, o “sesgada”, más en una dirección que en otra? Esa pequeña asimetría revela dónde la barrera cercana es más baja y en qué dirección es más probable que el sistema se escape.
Convertir la asimetría en un volante de dirección
El método comienza con una breve simulación no sesgada del sistema, durante la cual recoge muchas instantáneas de las posiciones atómicas y las convierte en descriptores numéricos, como las distancias de enlace. Dado que los sistemas químicos reales implican muchos de estos descriptores, los autores construyen una red neuronal especial llamada Skewencoder. Esta red comprime los datos de alta dimensión en una única coordenada que captura el movimiento lento esencial del sistema. De forma crucial, Skewencoder se entrena no solo para reconstruir los datos de entrada, sino también para maximizar la sesgadez —una medida de asimetría— de la distribución a lo largo de esta nueva coordenada. Una vez identificada esta dirección sesgada, Loxodynamics añade una suave “pared” armónica unidireccional que empuja el sistema más a lo largo de la dirección donde la cola de la distribución es más larga, orientándolo efectivamente cuesta arriba sobre la barrera cercana más baja.
Búsqueda iterativa sin conjeturas químicas
Loxodynamics procede en ciclos de “muestreo y búsqueda”. Tras cada simulación sesgada, la nueva trayectoria se añade a un conjunto de datos global en crecimiento usado para refinar la red neuronal, mientras que el último segmento se emplea para actualizar la guía basada en la sesgadez. Entonces se reposiciona la pared de sesgo según la nueva distribución sesgada y se vuelve a empujar el sistema. Este proceso iterativo lleva gradualmente al sistema fuera de su valle inicial y hacia nuevos estados metaestables que corresponden a intermedios o productos. Dado que la coordenada guía y la dirección se aprenden en tiempo real a partir de los datos, el método no requiere que el usuario especifique de antemano qué enlaces se romperán o formarán, ni qué productos esperar.

Probar el método desde modelos simples hasta catalizadores en funcionamiento
Para demostrar su potencia, los autores aplican primero Loxodynamics a paisajes modelo unidimensionales y bidimensionales sencillos, donde dos rutas diferentes conectan los mismos estados inicial y final. El método selecciona de forma fiable la ruta de menor barrera. Luego pasan a reacciones reales en fase gaseosa: una clásica reacción de sustitución (SN2) y una cicloaddición Diels–Alder. Incluso alimentado solo con descriptores de distancias generales, Loxodynamics encuentra automáticamente las vías de reacción correctas en ambas direcciones, afrontando regiones de reactivos anchas y flexibles y pozos de producto estrechos. Finalmente, el enfoque se prueba en escenarios catalíticos exigentes: deshidratación de etanol y 1‑butanol dentro de un marco zeolítico ácido. A temperaturas de operación realistas, el método descubre tanto mecanismos conocidos “concertados” como “paso a paso”, identifica intermedios de corta vida y distingue entre distintos productos alquénicos —todo ello usando descriptores simples basados en distancias y con mínima intervención humana.
Qué significa esto para el futuro del descubrimiento de reacciones
Este trabajo muestra que una idea relativamente simple —usar la sesgadez del movimiento local para apuntar hacia la ruta de escape más fácil— puede convertirse en una herramienta práctica de orientación para simulaciones moleculares. Al combinar dinámica sesgada con una red neuronal consciente de la sesgadez, Loxodynamics ofrece una vía sistemática para descubrir canales de reacción e intermedios probables a temperatura finita, sin elevar artificialmente la temperatura ni predefinir coordenadas de reacción detalladas. A largo plazo, tales enfoques podrían acelerar el diseño de catalizadores, mejorar los potenciales interatómicos aprendidos por máquina en las regiones de transición difíciles de muestrear e incluso adaptarse a problemas no químicos como el plegamiento de proteínas o la nucleación de cristales, siempre que los sistemas deban encontrar rutas raras fuera de paisajes de energía complejos.
Cita: Zhang, Z., Piccini, G. Exploring chemistry and catalysis by biasing skewed distributions via deep learning. Nat Commun 17, 3010 (2026). https://doi.org/10.1038/s41467-026-69586-8
Palabras clave: descubrimiento de reacciones, dinámica molecular, catálisis, aprendizaje profundo, paisajes de energía libre