Clear Sky Science · it
Esplorare la chimica e la catalisi dirigendo distribuzioni asimmetriche tramite deep learning
Perché questo nuovo strumento chimico è importante
I chimici vogliono sapere non solo come avvengono le reazioni, ma quali reazioni sono effettivamente possibili quando le molecole si incontrano in condizioni reali—in solventi, su superfici catalitiche, a temperature operative. Tradizionalmente, mappare tutte le possibili riorganizzazioni degli atomi richiede ipotesi laboriose e calcoli intensivi. Questo articolo presenta un nuovo approccio computazionale, chiamato Loxodynamics, che sfrutta concetti di statistica e deep learning per spingere automaticamente le simulazioni molecolari verso i percorsi di reazione più probabili, senza necessitare dell’intuito umano a priori su quali prodotti si formeranno.
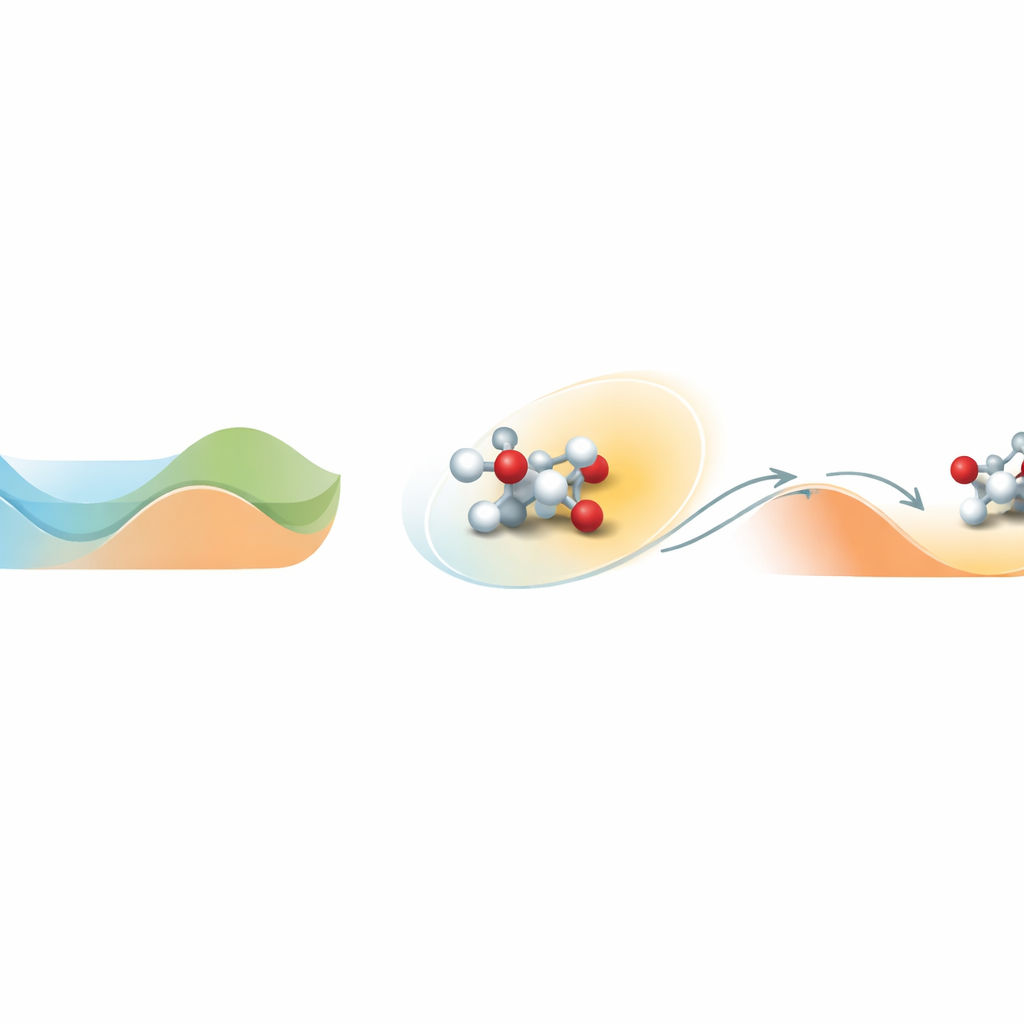
Trovare l’uscita da una valle energetica
Al centro del problema c’è il modo in cui le molecole si muovono su un paesaggio energetico complesso pieno di valli e colline. Una valle rappresenta uno stato stabile; salire una collina corrisponde a superare una barriera di reazione. Le simulazioni molecolari standard a temperature realistiche spesso restano intrappolate in una valle e non osservano mai gli eventi rari di attraversamento della barriera che definiscono la chimica. Loxodynamics affronta questo osservando come un sistema vaga all’interno di una valle e ponendo una semplice domanda statistica: la nuvola locale di posizioni è leggermente allungata, o “skewata”, più in una direzione che in un’altra? Questa piccola asimmetria rivela dove la barriera vicina è più bassa e in quale direzione il sistema è più probabile che sfugga.
Trasformare l’asimmetria in un volante di guida
Il metodo inizia con una breve simulazione non biasata del sistema, durante la quale raccoglie molti istantanee delle posizioni atomiche e le converte in descrittori numerici, come distanze di legame. Poiché i sistemi chimici reali coinvolgono molti di questi descrittori, gli autori costruiscono una rete neurale speciale chiamata Skewencoder. Questa rete comprime i dati ad alta dimensione in una singola coordinata che cattura il moto lento essenziale del sistema. Fondamentalmente, lo Skewencoder viene addestrato non solo a ricostruire i dati in ingresso, ma anche a massimizzare la skewness—una misura di asimmetria—della distribuzione lungo questa nuova coordinata. Una volta individuata questa direzione skewata, Loxodynamics aggiunge una “muro” armonico unilaterale e delicato che spinge il sistema ulteriormente lungo la direzione dove la coda della distribuzione è più lunga, guidandolo efficacemente in salita oltre la barriera vicina più bassa.
Ricerca iterativa senza congetture chimiche
Loxodynamics procede in cicli di “campionamento e ricerca”. Dopo ogni simulazione biasata, la nuova traiettoria viene aggiunta a un dataset globale in crescita usato per affinare la rete neurale, mentre il segmento più recente viene usato per aggiornare la guida basata sulla skewness. Il muro di bias viene quindi riposizionato in base alla nuova distribuzione skewata e il sistema viene spinto di nuovo. Questo processo iterativo porta gradualmente il sistema fuori dalla valle iniziale e verso nuovi stati metastabili che corrispondono a intermedi o prodotti. Poiché la coordinata guida e la direzione sono imparate al volo dai dati, il metodo non richiede che l’utente specifichi in anticipo quali legami si romperanno o si formeranno, o quali prodotti aspettarsi.

Testare il metodo da modelli semplici a catalizzatori operativi
Per dimostrarne l’efficacia, gli autori applicano prima Loxodynamics a modelli unidimensionali e bidimensionali semplici, dove due percorsi differenti collegano gli stessi stati iniziale e finale. Il metodo individua in modo affidabile la via con la barriera più bassa. Passano poi a reazioni in fase gassosa reali: una classica reazione di sostituzione (SN2) e una cicladdizione di Diels–Alder. Anche alimentato solo con descrittori di distanza generici, Loxodynamics trova automaticamente i corretti percorsi di reazione sia in avanti sia all’indietro, gestendo regioni di reagenti ampie e flessibili e pozzi di prodotti stretti. Infine, l’approccio viene testato in scenari catalitici impegnativi: disidratazione di etanolo e 1‑butanolo all’interno di una struttura zeolitica acida. A temperature operative realistiche, il metodo rivela sia meccanismi noti “concertati” sia “a gradini”, identifica intermedi di vita breve e distingue tra diversi prodotti alchenici—tutto usando descrittori semplici basati sulle distanze e con un intervento umano minimo.
Cosa significa per la scoperta futura di reazioni
Questo lavoro mostra che un’idea relativamente semplice—usare la skewness del moto locale per indicare la via di fuga più facile—può essere trasformata in uno strumento pratico di guida per le simulazioni molecolari. Combinando dinamiche biasate con una rete neurale sensibile alla skewness, Loxodynamics offre un modo sistematico per scoprire canali di reazione e intermedi probabili a temperatura finita, senza aumentare artificialmente la temperatura o predefinire coordinate di reazione dettagliate. A lungo termine, tali approcci potrebbero accelerare la progettazione di catalizzatori, migliorare i potenziali interatomici appresi dalle macchine nelle regioni di transizione difficili da campionare e persino essere adattati a problemi non chimici come il ripiegamento proteico o la nucleazione cristallina, ovunque i sistemi debbano trovare percorsi rari per uscire da paesaggi energetici complessi.
Citazione: Zhang, Z., Piccini, G. Exploring chemistry and catalysis by biasing skewed distributions via deep learning. Nat Commun 17, 3010 (2026). https://doi.org/10.1038/s41467-026-69586-8
Parole chiave: scoperta di reazioni, dinamica molecolare, catalisi, deep learning, paesaggi di energia libera