Clear Sky Science · de
Chemie und Katalyse erkunden durch Verzerrung schiefer Verteilungen mithilfe von Deep Learning
Warum dieses neue Chemie-Werkzeug wichtig ist
Chemiker möchten nicht nur verstehen, wie Reaktionen ablaufen, sondern auch, welche Reaktionen überhaupt möglich sind, wenn Moleküle unter realen Bedingungen zusammentreffen – in Lösungsmitteln, auf Katalysatoroberflächen, bei Arbeitstemperaturen. Traditionell erfordert das vollständige Aufspüren aller möglichen Umordnungen von Atomen mühsame Vermutungen und aufwändige Rechnungen. Dieses Paper stellt einen neuen rechnergestützten Ansatz vor, genannt Loxodynamics, der Konzepte aus Statistik und Deep Learning nutzt, um Molekülsimulationen automatisch in Richtung der wahrscheinlichsten Reaktionspfade zu lenken, ohne dass zuvor menschliche Intuition über zu bildende Produkte nötig ist.
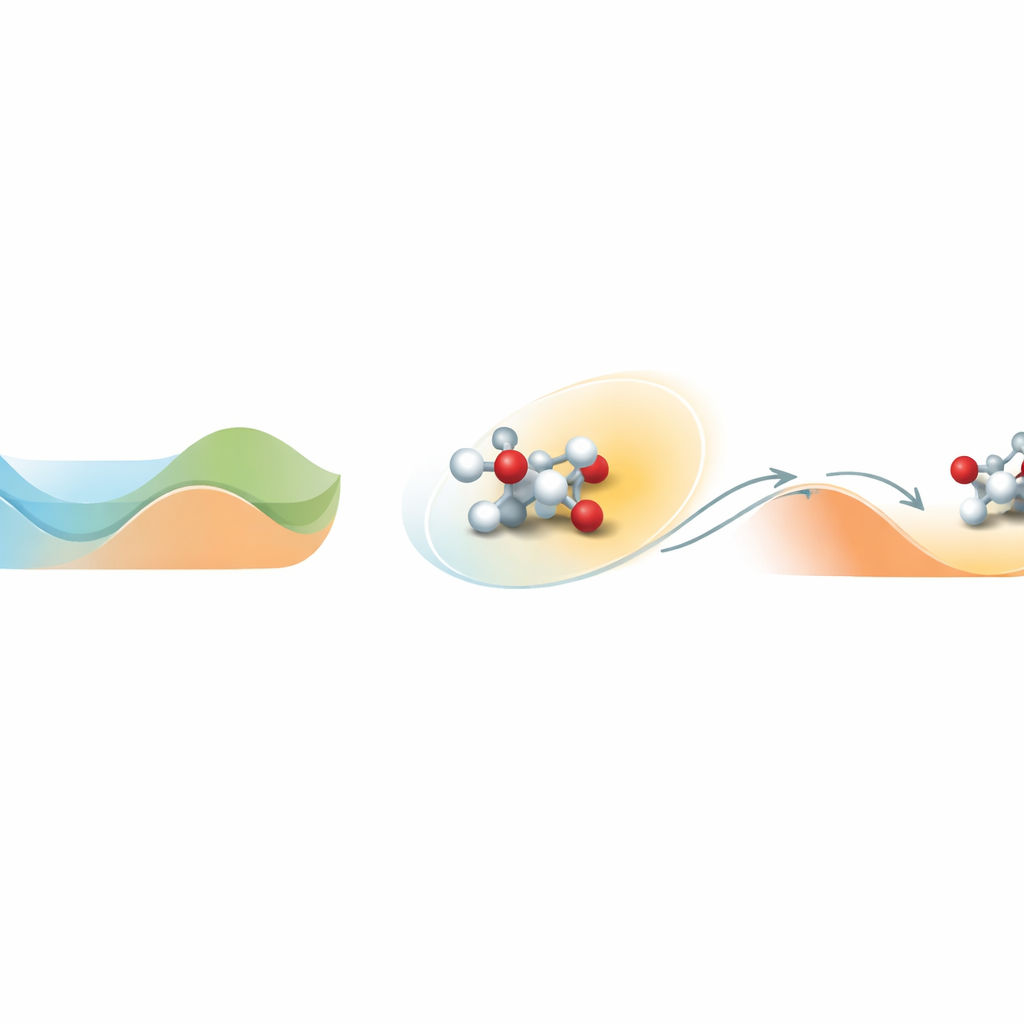
Den Weg aus einem Energie-Tal finden
Im Kern des Problems steht, wie sich Moleküle auf einer komplexen Energielandschaft mit Tälern und Hügeln bewegen. Ein Tal steht für einen stabilen Zustand; das Überwinden eines Hügels entspricht dem Überschreiten einer Reaktionsbarriere. Standard-Molekülsimulationen bei realistischen Temperaturen bleiben oft in einem Tal gefangen und beobachten nie die seltenen Barrieredurchbrüche, die Chemie ausmachen. Loxodynamics begegnet dem, indem es verfolgt, wie ein System innerhalb eines Tals wandert, und eine einfache statistische Frage stellt: Ist die lokale Wolke von Positionen in eine Richtung leicht gestreckt oder „schief“? Diese kleine Asymmetrie verrät, wo die nächste Barriere am niedrigsten ist und in welche Richtung das System am wahrscheinlichsten entkommen wird.
Asymmetrie als Lenkrad einsetzen
Die Methode beginnt mit einer kurzen, unverfälschten Simulation des Systems, während der viele Momentaufnahmen atomarer Positionen gesammelt und in numerische Beschreibungen wie Bindungsabstände umgewandelt werden. Da reale chemische Systeme viele solche Deskriptoren enthalten, bauen die Autoren ein spezielles neuronales Netz namens Skewencoder. Dieses Netz komprimiert die hochdimensionale Datenmenge zu einer einzigen Koordinate, die die wesentliche langsame Bewegung des Systems erfasst. Entscheidend ist, dass Skewencoder nicht nur darauf trainiert wird, die Eingangsdaten zu rekonstruieren, sondern auch die Schiefe – ein Maß für Asymmetrie – der Verteilung entlang dieser neuen Koordinate zu maximieren. Sobald diese schiefe Richtung identifiziert ist, fügt Loxodynamics eine sanfte, einseitige harmonische „Wand“ hinzu, die das System weiter in die Richtung schiebt, in der der Verteilungsschwanz länger ist, und es so effektiv bergauf über die niedrigste nahegelegene Barriere steuert.
Iteratives Suchen ohne chemische Vorannahmen
Loxodynamics arbeitet in Zyklen von „Stichproben nehmen und suchen“. Nach jeder biasierten Simulation wird die neue Trajektorie zu einem wachsenden globalen Datensatz hinzugefügt, der das neuronale Netz verfeinert, während das jüngste Segment genutzt wird, um die schiefheitsbasierte Steuerung zu aktualisieren. Die Bias-Wand wird dann anhand der neuen schiefen Verteilung neu positioniert und das System erneut angestoßen. Dieser iterative Prozess führt das System schrittweise aus seinem Anfangstal in neue metastabile Zustände, die Zwischenprodukte oder Endprodukte entsprechen. Da die leitende Koordinate und Richtung on-the-fly aus den Daten erlernt werden, muss der Anwender nicht im Voraus angeben, welche Bindungen brechen oder entstehen oder welche Produkte zu erwarten sind.

Methodenprüfung von einfachen Modellen bis zu arbeitenden Katalysatoren
Um die Leistungsfähigkeit zu demonstrieren, wenden die Autoren Loxodynamics zunächst auf einfache ein- und zweidimensionale Modelllandschaften an, bei denen zwei unterschiedliche Pfade denselben Start- und Endzustand verbinden. Die Methode erkennt zuverlässig die Route mit der niedrigeren Barriere. Anschließend gehen sie zu realen Gasphasenreaktionen über: einer klassischen Substitutionsreaktion (SN2) und einer Diels–Alder-Cycloaddition. Selbst wenn nur allgemeine Distanzdeskriptoren verwendet werden, findet Loxodynamics automatisch die korrekten Reaktionspfade in Vorwärts- und Rückwärtsrichtung und meistert dabei breite, flexible Reaktantenbereiche und enge Produktmulden. Schließlich wird der Ansatz in anspruchsvollen katalytischen Szenarien getestet: Dehydratisierung von Ethanol und 1‑Butanol innerhalb eines saueren Zeolith-Rahmens. Bei realistischen Betriebstemperaturen deckt die Methode sowohl bekannte „konzertierte“ als auch „schrittweise“ Mechanismen auf, identifiziert kurzlebige Zwischenstufen und unterscheidet zwischen verschiedenen Alkenprodukten – und das alles bei Verwendung einfacher distanzbasierter Deskriptoren und minimalem menschlichen Eingriff.
Was das für die künftige Reaktionsentdeckung bedeutet
Diese Arbeit zeigt, dass eine relativ einfache Idee – die Schiefe lokaler Bewegung zu nutzen, um auf den leichtesten Ausweg zu zeigen – zu einem praktischen Steuerungswerkzeug für Molekülsimulationen gemacht werden kann. Durch die Kombination biasierter Dynamik mit einem schiefheitsbewussten neuronalen Netz bietet Loxodynamics einen systematischen Weg, wahrscheinliche Reaktionskanäle und Zwischenstufen bei endlicher Temperatur zu entdecken, ohne die Temperatur künstlich zu erhöhen oder detaillierte Reaktionskoordinaten vorzugeben. Langfristig könnten solche Ansätze das Design von Katalysatoren beschleunigen, maschinell gelernte interatomare Potentiale in schwer zu sammelnden Übergangsregionen verbessern und sogar auf nicht-chemische Probleme wie Protein-Faltung oder Kristallkeimbildung übertragen werden – überall dort, wo Systeme seltene Pfade aus komplexen Energielandschaften finden müssen.
Zitation: Zhang, Z., Piccini, G. Exploring chemistry and catalysis by biasing skewed distributions via deep learning. Nat Commun 17, 3010 (2026). https://doi.org/10.1038/s41467-026-69586-8
Schlüsselwörter: Reaktionsentdeckung, Molekulardynamik, Katalyse, Deep Learning, Freienergie-Landschaften