Clear Sky Science · en
Exploring chemistry and catalysis by biasing skewed distributions via deep learning
Why this new chemistry tool matters
Chemists want to know not just how reactions work, but what reactions are even possible when molecules meet under real-world conditions—inside solvents, on catalyst surfaces, at working temperatures. Traditionally, mapping out all the ways atoms can rearrange has required painstaking guesswork and heavy calculations. This paper introduces a new computer-based approach, called Loxodynamics, that uses ideas from statistics and deep learning to automatically push molecular simulations toward the most likely reaction pathways, without needing prior human intuition about what products will form.
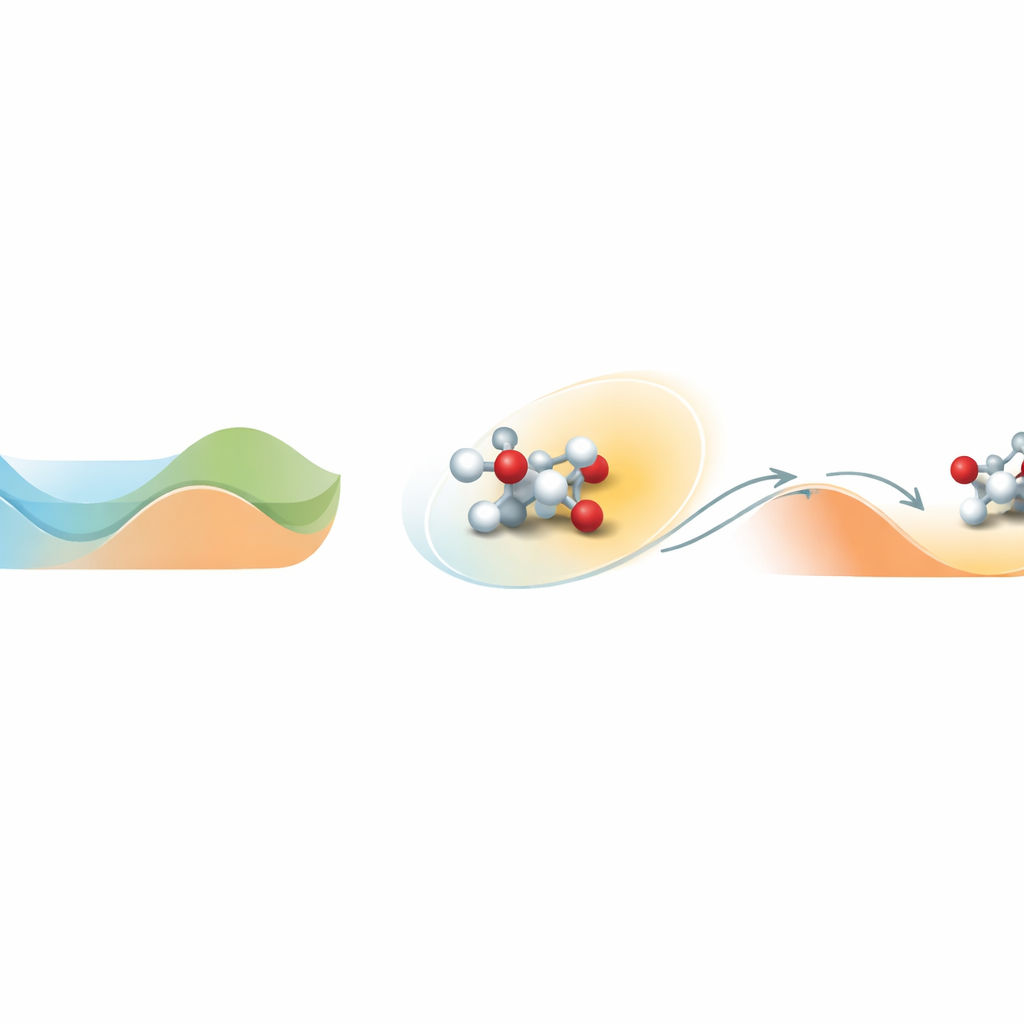
Finding the way out of an energy valley
At the heart of the problem is how molecules move on a complex energy landscape full of valleys and hills. A valley represents a stable state; climbing a hill corresponds to crossing a reaction barrier. Standard molecular simulations at realistic temperatures often get stuck in one valley and never see the rare barrier-crossing events that define chemistry. Loxodynamics tackles this by watching how a system wanders within a valley and asking a simple statistical question: is the local cloud of positions slightly stretched, or “skewed,” more in one direction than another? That small asymmetry reveals where the nearby barrier is lowest and in which direction the system is most likely to escape.
Turning asymmetry into a steering wheel
The method starts with a brief, unbiased simulation of the system, during which it collects many snapshots of atomic positions and converts them into numerical descriptors, such as bond distances. Because real chemical systems involve many such descriptors, the authors build a special neural network called Skewencoder. This network compresses the high-dimensional data into a single coordinate that captures the essential slow motion of the system. Crucially, Skewencoder is trained not only to reconstruct the input data, but also to maximize the skewness—an asymmetry measure—of the distribution along this new coordinate. Once this skewed direction is identified, Loxodynamics adds a gentle, one-sided harmonic “wall” that nudges the system further along the direction where the tail of the distribution is longer, effectively steering it uphill over the lowest nearby barrier.
Iterative searching without chemical guesswork
Loxodynamics proceeds in cycles of “sample and search.” After each biased simulation, the new trajectory is added to a growing global dataset used to refine the neural network, while the latest segment is used to update the skewness-based guidance. The biasing wall is then repositioned based on the new skewed distribution, and the system is pushed again. This iterative process gradually leads the system out of its initial valley and into new metastable states that correspond to intermediates or products. Because the guiding coordinate and direction are learned on the fly from the data, the method does not require the user to specify in advance which bonds will break or form, or which products to expect.

Testing the method from simple models to working catalysts
To demonstrate its power, the authors first apply Loxodynamics to simple one- and two-dimensional model landscapes, where two different paths connect the same starting and final states. The method reliably picks out the lower barrier route. They then move to real gas-phase reactions: a classic substitution reaction (SN2) and a Diels–Alder cycloaddition. Even when fed only general distance descriptors, Loxodynamics automatically finds the correct reaction pathways in both forward and reverse directions, coping with broad, floppy reactant regions and narrow product wells. Finally, the approach is tested in demanding catalytic scenarios: dehydration of ethanol and 1‑butanol inside an acidic zeolite framework. Under realistic operating temperatures, the method uncovers both known “concerted” and “stepwise” mechanisms, identifies short-lived intermediates, and distinguishes between different alkene products—all while using simple distance-based descriptors and minimal human input.
What this means for future reaction discovery
This work shows that a relatively simple idea—using the skewness of local motion to point toward the easiest escape route—can be turned into a practical steering tool for molecular simulations. By combining biased dynamics with a skewness-aware neural network, Loxodynamics offers a systematic way to discover likely reaction channels and intermediates at finite temperature, without raising the temperature artificially or predefining detailed reaction coordinates. In the long term, such approaches could accelerate the design of catalysts, improve machine-learned interatomic potentials in the hard-to-sample transition regions, and even be adapted to non-chemical problems like protein folding or crystal nucleation, wherever systems must find rare pathways out of complex energy landscapes.
Citation: Zhang, Z., Piccini, G. Exploring chemistry and catalysis by biasing skewed distributions via deep learning. Nat Commun 17, 3010 (2026). https://doi.org/10.1038/s41467-026-69586-8
Keywords: reaction discovery, molecular dynamics, catalysis, deep learning, free energy landscapes