Clear Sky Science · pl
AKT1 fosforyluje PRMT7, promując metylację GLUD1 i rozwój raka żołądka
Dlaczego to ma znaczenie dla leczenia raka
Komórki nowotworowe są znane ze swego ogromnego apetytu, przebudowując metabolizm, by rosnąć i się rozprzestrzeniać. Badanie to odkrywa wcześniej nieznany obwód molekularny, który pomaga komórkom raka żołądka korzystać z substancji odżywczej — glutaminy — gdy zapasy cukru się wahają. Odszyfrowując, jak ten układ utrzymuje przy życiu i aktywną kluczową enzymatyczną protezę metaboliczną, autorzy ujawniają nowe słabe ogniwo, które mogłoby uczynić istniejące chemioterapeutyki bardziej skutecznymi przeciw rakowi żołądka.
Przełącznik paliwa, który pomaga guzom przetrwać
Większość osób słyszała o zamiłowaniu raka do cukru, często opisywanym jako efekt Warburga. Ale komórki nowotworowe mogą też silnie polegać na glutaminie, obfitym aminokwasie, który zasila ich „fabryki energii” i dostarcza cegiełek do tworzenia DNA oraz innych cząsteczek. W centrum tego procesu znajduje się enzym GLUD1, który przekształca glutaminian pochodzący z glutaminy w związek zasila cykl energetyczny komórki. GLUD1 jest często podwyższony w wielu nowotworach, w tym w guzach żołądka; wcześniejsze prace wykazały, że blokowanie GLUD1 może spowalniać wzrost guza i wzmacniać działanie chemioterapii. Do tej pory naukowcy nie rozumieli jednak, jak komórki nowotworowe precyzyjnie regulują ilość i aktywność GLUD1 w odpowiedzi na zmiany dostępności składników odżywczych.
Chemiczny znacznik, który chroni kluczowy enzym
Autorzy odkryli, że GLUD1 jest kontrolowany przez mały chemiczny znacznik na jednym z jego aminokwasów — argininie w pozycji 76. Ten znacznik, grupa metylowa, jest dodawany przez enzym PRMT7. Przy użyciu przeciwciała zaprojektowanego tak, by rozpoznawać GLUD1 tylko wtedy, gdy ta arginina jest metylowana, pokazali, że to miejsce stanowi główną i silnie zachowaną modyfikację. Gdy metylacja była blokowana farmakologicznie lub przez mutację tej argininy uniemożliwiającą modyfikację, poziomy GLUD1 szybko spadały, mimo że jego zapis genetyczny pozostawał niezmieniony. Powód: bez metylacji GLUD1 był bardziej oznaczany ubikwityną — sygnałem kierującym białka do komórkowego systemu utylizacji. Innymi słowy, metylacja działa jak tarcza ochronna, która zapobiega rozkładowi GLUD1.
Jak poziom cukru we krwi i sygnały wzrostu się z tym wiążą
Zespół następnie zbadał, jak warunki odżywcze poza komórką wpływają na tę ochronną metylację wewnątrz komórki. Stwierdzili, że wysokie stężenia glukozy obniżały poziom białka GLUD1, podczas gdy niski poziom glukozy, insulina lub lek przeciwcukrzycowy metformina sprzyjały akumulacji GLUD1. Efekty te odprowadzały do ścieżki PI3K/Akt, głównego szlaku sygnalizacyjnego kontrolującego odpowiedź komórek na czynniki wzrostu i wykorzystanie glukozy. Gdy ten szlak, a zwłaszcza enzym AKT1, był hamowany, GLUD1 stawał się mniej metylowany i bardziej ubikwitynowany, co prowadziło do jego degradacji. Dalsze eksperymenty wykazały, że AKT1 nie modyfikuje GLUD1 bezpośrednio; zamiast tego wiąże się z PRMT7 i fosforyluje go w określonym treoninie (T73). Ta fosforylacja zwiększa aktywność PRMT7 i jego zdolność do metylowania GLUD1, stabilizując tym samym GLUD1 i utrzymując metabolizm glutaminy podczas stresu związanego z niską glukozą.
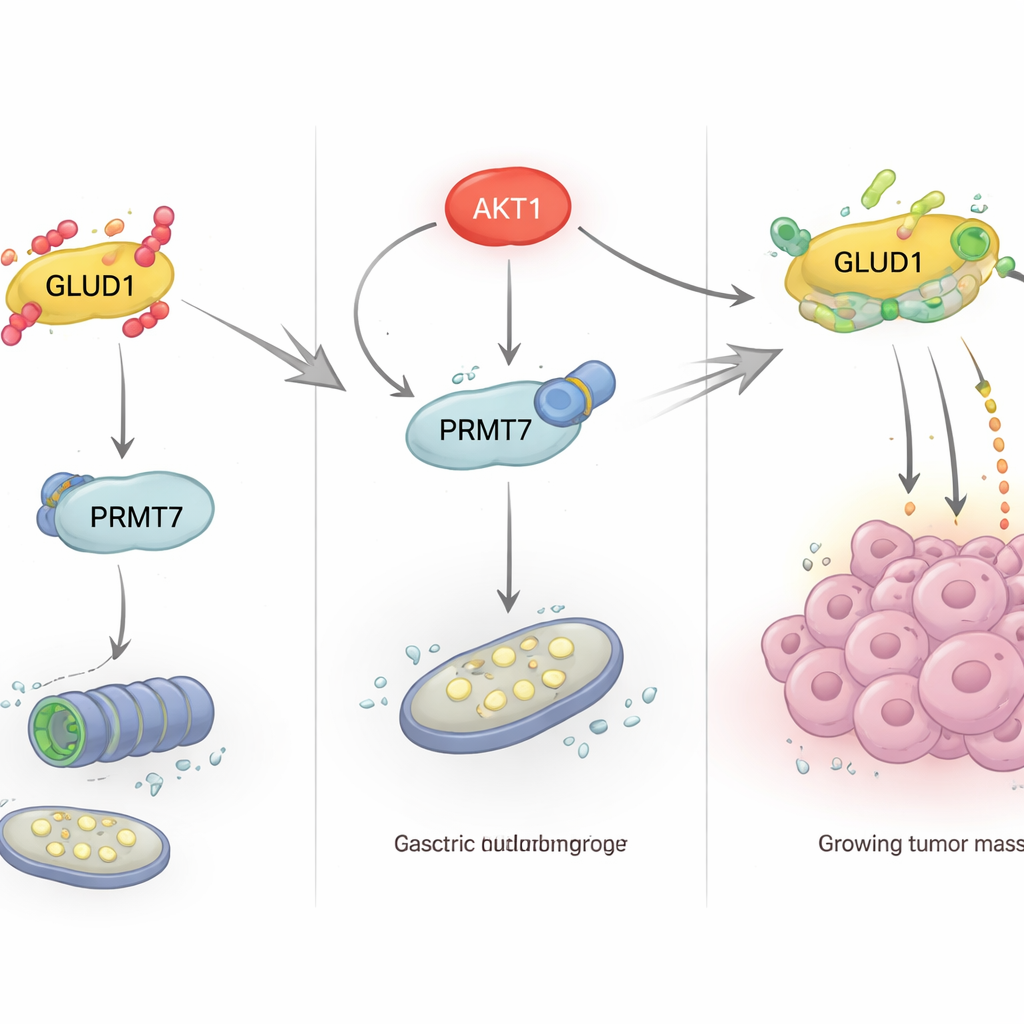
Od molekularnego przełącznika do wzrostu guza
Stabilizacja GLUD1 to nie tylko biochemiczna ciekawostka — ma wymierne konsekwencje dla zachowania guza. Gdy badacze zastąpili normalny GLUD1 wersją pozbawioną możliwości metylacji w komórkach raka żołądka, aktywność enzymatyczna GLUD1 spadła, produkcja energii i prekursorów napędzana przez glutaminę zmniejszyła się, a komórki proliferowały i migrowały słabiej. Zablokowanie PRMT7 za pomocą narzędzi genetycznych lub selektywnego leku SGC3027 dało podobny efekt: obniżenie metylacji GLUD1, spadek poziomu białka GLUD1 oraz spowolnienie wzrostu i ruchu komórek nowotworowych. W próbkach tkankowych od pacjentów z rakiem żołądka guzy wykazywały wyższe poziomy GLUD1, metylowanego GLUD1 i PRMT7 niż otaczająca tkanka normalna, a te markery zmieniały się synchronicznie, co wspiera tezę, że ta oś jest aktywna w chorobie ludzkiej.
Nowa nadzieja na wzmocnienie chemioterapii
Ponieważ GLUD1 był już powiązany z opornością na lek chemioterapeutyczny doksetaksel (DTX), autorzy przetestowali, czy ukierunkowanie na PRMT7 może poprawić terapię. W hodowlach komórkowych SGC3027 spowolnił wzrost komórek raka żołądka, a połączenie z DTX dało silniejsze zahamowanie niż każdy z leków osobno. Guzy hodowane u myszy kurczyły się znacznie bardziej, gdy oba leki podawano razem — około 80% spadek masy guza w porównaniu z kontrolami oraz wyraźne zmniejszenie markera podziałów komórkowych. Co istotne, guzy i linie komórkowe oporne na DTX wykazywały wyższą metylację GLUD1, co sugeruje, że blokada tej modyfikacji może pomóc przezwyciężyć oporność. W prostych słowach, badanie identyfikuje przekaźnik AKT1–PRMT7–GLUD1, który pozwala komórkom raka żołądka dostosować wykorzystanie paliwa i przetrwać, oraz pokazuje, że przecięcie tego przekaźnika może osłabić guzy i zwiększyć skuteczność istniejącej chemioterapii.
Cytowanie: Cui, Z., Li, H., Liang, X. et al. AKT1 phosphorylates PRMT7 to promote GLUD1 methylation and gastric cancer progression. Cell Death Dis 17, 363 (2026). https://doi.org/10.1038/s41419-026-08601-8
Słowa kluczowe: rak żołądka, metabolizm guza, glutamina, metylacja białek, terapia celowana