Clear Sky Science · de
AKT1 phosphoryliert PRMT7, um GLUD1-Methylierung und das Fortschreiten von Magenkrebs zu fördern
Warum das für die Krebsbehandlung wichtig ist
Krebszellen sind berüchtigt dafür, besonders viel Energie zu benötigen und ihren Stoffwechsel umzubauen, um zu wachsen und zu streuen. Diese Studie enthüllt einen zuvor verborgenen molekularen Regelkreis, der Magenkrebszellen hilft, auf das Nährstoffglutamin zurückzugreifen, wenn die Zuckerzufuhr schwankt. Indem die Forschenden entschlüsseln, wie dieser Mechanismus ein zentrales Stoffwechselenzym am Leben und aktiv hält, zeigen sie eine neue Verwundbarkeit auf, die bestehende Chemotherapien gegen Magenkrebs wirksamer machen könnte.
Ein Brennstoffwechsel-Umschalter, der Tumoren überleben lässt
Die meisten Menschen haben von der Vorliebe des Krebses für Zucker gehört, oft als Warburg-Effekt bezeichnet. Tumorzellen können sich aber auch stark auf Glutamin stützen, eine reichlich vorhandene Aminosäure, die ihre Energiezentralen versorgt und Bausteine für DNA und andere Moleküle liefert. Im Zentrum dieses Prozesses steht ein Enzym namens GLUD1, das aus Glutamin stammendes Glutamat in ein Molekül umwandelt, das in den zentralen Energiestoffwechsel eingespeist wird. GLUD1 ist in vielen Krebsarten, einschließlich MagenTumoren, häufig erhöht, und frühere Arbeiten zeigten, dass die Hemmung von GLUD1 das Tumorwachstum verlangsamen und Chemotherapien verstärken kann. Bislang war jedoch unklar, wie Krebszellen Menge und Aktivität von GLUD1 an wechselnde Nährstoffbedingungen anpassen.
Ein chemisches Kennzeichen, das ein Schlüsselenzym schützt
Die Autorinnen und Autoren entdeckten, dass GLUD1 durch eine kleine chemische Markierung an einer seiner Aminosäuren gesteuert wird: Arginin an Position 76. Diese Markierung, eine Methylgruppe, wird von einem Enzym namens PRMT7 angefügt. Mit einem speziell hergestellten Antikörper, der GLUD1 nur erkennt, wenn dieses Arginin methyliert ist, zeigten sie, dass diese Stelle eine bedeutende und hochkonservierte Modifikation darstellt. Wenn die Methylierung durch Medikamente blockiert oder durch Mutation dieses Arginins verhindert wurde, sanken die GLUD1-Proteinniveaus schnell, obwohl die genetische Vorlage unverändert blieb. Der Grund: Ohne Methylierung wurde GLUD1 stärker mit Ubiquitin markiert, ein Signal, das Proteine zur zellulären Abbau-Maschinerie schickt. Anders gesagt wirkt Methylierung wie ein Schutzschild, das GLUD1 davor bewahrt, zersetzt zu werden.
Wie Blutzucker und Wachstumssignale zusammenhängen
Das Team untersuchte anschließend, wie äußere Nährstoffbedingungen diese schützende Methylierung im Inneren beeinflussen. Sie stellten fest, dass hohe Glukosewerte GLUD1-Protein reduzierten, während niedrige Glukose, Insulin oder das Diabetesmedikament Metformin die Akkumulation von GLUD1 förderten. Diese Effekte ließen sich auf den PI3K/Akt-Signalweg zurückführen, eine zentrale Route, die steuert, wie Zellen auf Wachstumsfaktoren reagieren und Glukose nutzen. Wenn dieser Weg, insbesondere das Enzym AKT1, gehemmt wurde, war GLUD1 weniger methyliert und stärker ubiquitiniert, was seinen Abbau vorantrieb. Weitere Experimente zeigten, dass AKT1 GLUD1 nicht direkt verändert; stattdessen bindet es an PRMT7 und phosphoryliert dieses bei einem spezifischen Threonin (T73). Diese Phosphorylierung steigert die Aktivität von PRMT7 und dessen Fähigkeit, GLUD1 zu methylieren, wodurch GLUD1 stabilisiert wird und der Glutaminstoffwechsel bei niedrigem Glukosespiegeldruck aufrechterhalten bleibt.
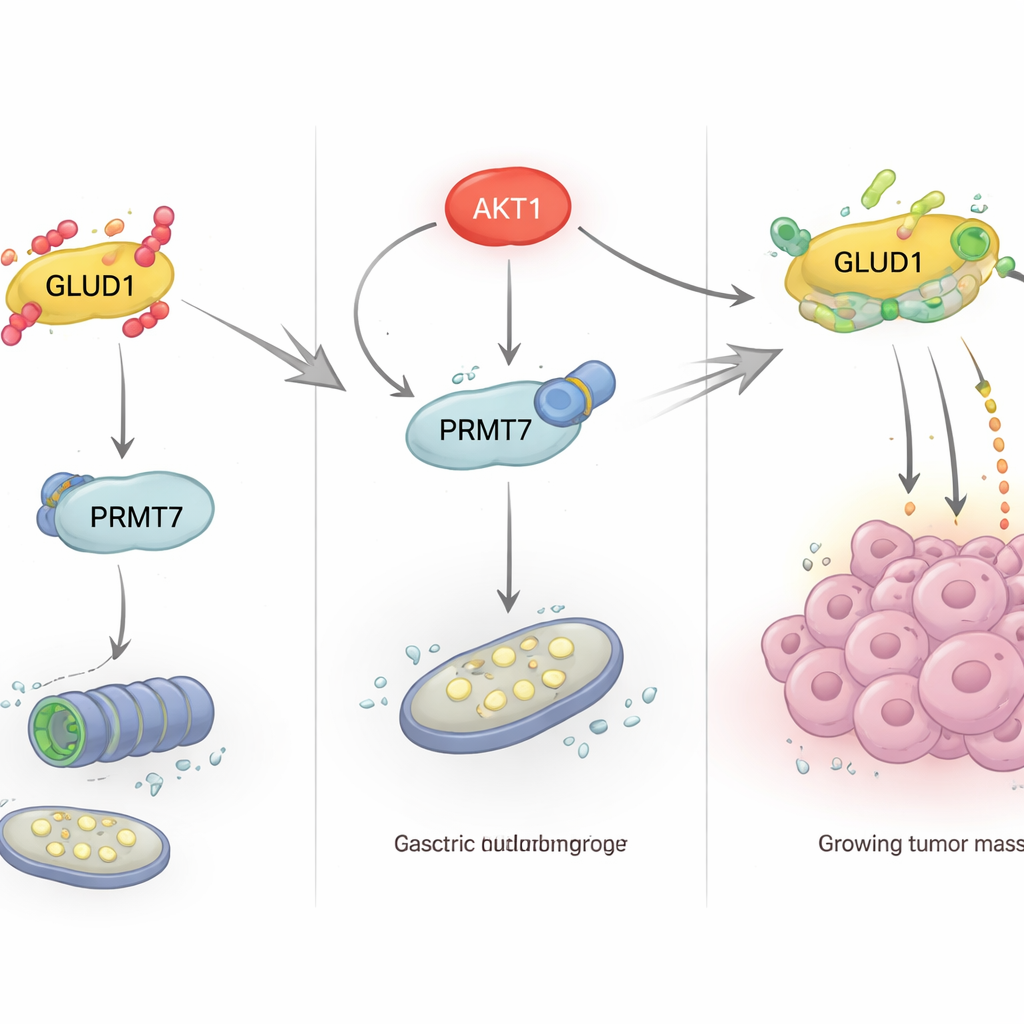
Vom molekularen Schalter zum Tumorwachstum
Die Stabilisierung von GLUD1 ist nicht nur eine biochemische Kuriosität; sie hat greifbare Folgen für das Tumorverhalten. Wenn die Forschenden das normale GLUD1 in Magenkrebszellen durch eine methylierungsdefekte Variante ersetzten, sank die Enzymaktivität von GLUD1, die glutamingetriebene Energie- und Baustoffproduktion ging zurück, und die Zellen proliferierten und migrierten weniger. Die Blockade von PRMT7 mit genetischen Werkzeugen oder einem selektiven Wirkstoff, SGC3027, ahmte diesen Effekt nach: GLUD1-Methylierung wurde reduziert, GLUD1-Proteinlevels sanken, und das Wachstum sowie die Bewegung der Krebszellen verlangsamten sich. In Gewebeproben von Magenkrebspatienten zeigten Tumoren höhere Werte von GLUD1, methyliertem GLUD1 und PRMT7 als angrenzendes normales Gewebe, und diese Marker stiegen und fielen gemeinsam, was die Idee stützt, dass diese Achse beim Menschen aktiv ist.
Neue Hoffnung zur Verstärkung von Chemotherapie
Da GLUD1 bereits mit Resistenz gegen das Chemotherapeutikum Docetaxel (DTX) in Verbindung gebracht wurde, prüften die Autorinnen und Autoren, ob die Zielung von PRMT7 die Behandlung verbessern könnte. In Zellkultur verlangsamte SGC3027 das Wachstum von Magenkrebszellen, und die Kombination mit DTX führte zu stärkerer Hemmung als jeder Wirkstoff allein. In Mäusemodellen schrumpften Tumoren deutlich stärker, wenn beide Wirkstoffe zusammen verabreicht wurden, mit etwa 80 % geringerer Tumorlast im Vergleich zu unbehandelten Kontrollen und einer deutlichen Reduktion eines Teilungsmarkers. Bemerkenswerterweise zeigten Tumoren und Zelllinien, die gegen DTX resistent waren, höhere GLUD1-Methylierung, was darauf hindeutet, dass die Blockade dieser Modifikation helfen könnte, Resistenz zu überwinden. Kurz gesagt identifiziert die Studie ein AKT1–PRMT7–GLUD1-Relais, das Magenkrebszellen ermöglicht, ihren Brennstoffverbrauch anzupassen und zu gedeihen, und zeigt, dass das Unterbrechen dieses Relais Tumoren schwächen und die Wirkung bestehender Chemotherapien verstärken kann.
Zitation: Cui, Z., Li, H., Liang, X. et al. AKT1 phosphorylates PRMT7 to promote GLUD1 methylation and gastric cancer progression. Cell Death Dis 17, 363 (2026). https://doi.org/10.1038/s41419-026-08601-8
Schlüsselwörter: Magenkrebs, Tumorstoffwechsel, Glutamin, Proteinmethylierung, gezielte Therapie