Clear Sky Science · en
AKT1 phosphorylates PRMT7 to promote GLUD1 methylation and gastric cancer progression
Why this matters for cancer treatment
Cancer cells are notoriously hungry, rewiring their metabolism to grow and spread. This study uncovers a previously hidden molecular circuit that helps stomach cancer cells tap into the nutrient glutamine when sugar supplies fluctuate. By decoding how this circuit keeps a key metabolic enzyme alive and active, the researchers reveal a new weak spot that could make existing chemotherapy drugs more effective against gastric cancer.
A fuel switch that helps tumors survive
Most people have heard of cancer’s love of sugar, often described as the Warburg effect. But tumor cells can also lean heavily on glutamine, an abundant amino acid that feeds their energy factories and supplies building blocks for DNA and other molecules. At the center of this process sits an enzyme called GLUD1, which turns glutamine-derived glutamate into a molecule that feeds the cell’s main energy cycle. GLUD1 is frequently elevated in many cancers, including gastric tumors, and earlier work showed that blocking GLUD1 can slow tumor growth and make chemotherapy more potent. Until now, however, scientists did not understand how cancer cells fine-tune the amount and activity of GLUD1 as nutrient conditions change.
A chemical tag that protects a key enzyme
The authors discovered that GLUD1 is controlled by a small chemical tag on one of its building blocks, an amino acid called arginine at position 76. This tag, a methyl group, is added by an enzyme named PRMT7. Using a custom-made antibody that recognizes GLUD1 only when this arginine is methylated, they showed that this site is a major and highly conserved modification. When methylation was blocked with drugs or by mutating this arginine so it could no longer be modified, GLUD1 levels dropped quickly even though its genetic blueprint stayed the same. The reason: without methylation, GLUD1 was more heavily marked with ubiquitin, a signal that sends proteins to the cell’s disposal machinery. In other words, methylation acts like a protective shield that prevents GLUD1 from being shredded.
How blood sugar and growth signals tie in
The team then asked how nutrient conditions outside the cell influence this protective methylation inside. They found that high glucose levels reduced GLUD1 protein, while low glucose, insulin, or the diabetes drug metformin favored GLUD1 accumulation. These effects traced back to the PI3K/Akt pathway, a major signaling route that controls how cells respond to growth factors and use glucose. When this pathway, and especially the enzyme AKT1, was inhibited, GLUD1 became less methylated and more ubiquitinated, driving its degradation. Further experiments revealed that AKT1 does not modify GLUD1 directly; instead, it binds to and phosphorylates PRMT7 at a specific threonine (T73). This phosphorylation boosts PRMT7’s activity and ability to methylate GLUD1, thereby stabilizing GLUD1 and maintaining glutamine metabolism under low-glucose stress.
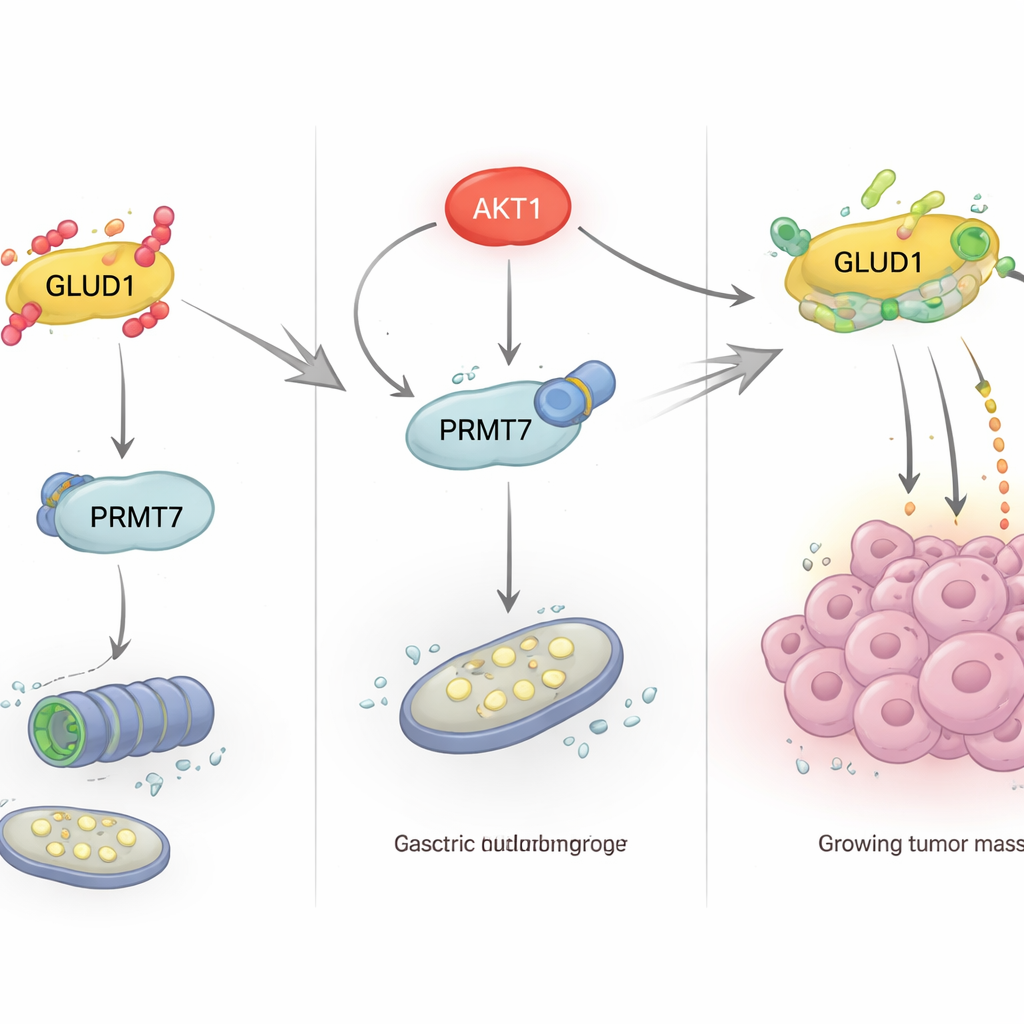
From molecular switch to tumor growth
Stabilizing GLUD1 is not just a biochemical curiosity; it has tangible consequences for tumor behavior. When the researchers replaced normal GLUD1 with a methylation-defective version in gastric cancer cells, GLUD1’s enzyme activity fell, glutamine-driven energy and building-block production declined, and cells proliferated and migrated less. Blocking PRMT7 with genetic tools or a selective drug, SGC3027, mimicked this effect, reducing GLUD1 methylation, lowering GLUD1 protein levels, and slowing cancer cell growth and movement. In tissue samples from gastric cancer patients, tumors showed higher levels of GLUD1, methylated GLUD1, and PRMT7 than nearby normal tissue, and these markers rose and fell together, supporting the idea that this axis is active in human disease.
New hope for boosting chemotherapy
Because GLUD1 has already been linked to resistance to the chemotherapy drug docetaxel (DTX), the authors tested whether targeting PRMT7 could improve treatment. In cell culture, SGC3027 slowed gastric cancer cell growth, and combining it with DTX produced stronger inhibition than either agent alone. Tumors grown in mice shrank far more when the two drugs were given together, with about an 80% drop in tumor weight compared to untreated controls and a marked reduction in a marker of cell division. Notably, tumors and cell lines resistant to DTX showed higher GLUD1 methylation, suggesting that blocking this modification could help overcome resistance. In simple terms, the study identifies an AKT1–PRMT7–GLUD1 relay that lets gastric cancer cells adjust their fuel use and thrive, and shows that cutting this relay can weaken tumors and sharpen the impact of existing chemotherapy.
Citation: Cui, Z., Li, H., Liang, X. et al. AKT1 phosphorylates PRMT7 to promote GLUD1 methylation and gastric cancer progression. Cell Death Dis 17, 363 (2026). https://doi.org/10.1038/s41419-026-08601-8
Keywords: gastric cancer, tumor metabolism, glutamine, protein methylation, targeted therapy