Clear Sky Science · en
Spatial transcriptomic landscape of invasion patterns in human papillomavirus-associated endocervical adenocarcinoma
Why this matters for women’s health
Cervical cancer is often thought of as a single disease, but cancers of the cervix do not all behave the same. This study looks closely at a form linked to human papillomavirus (HPV) called endocervical adenocarcinoma and asks a simple but crucial question: why do some tumors stay relatively contained while others invade deeply and become more dangerous? By mapping which genes are active in different parts of the tumor and its surrounding tissue, the authors uncover clues that could one day help doctors better predict risk and design more targeted treatments.
Different ways a cancer can spread
Doctors already know that HPV-associated endocervical adenocarcinomas can be grouped into patterns of invasion, known as Silva patterns A, B, and C. Pattern A tumors tend to grow in rounded glands and stay more organized, while pattern C tumors invade in a more destructive, diffuse way and are much more likely to spread to lymph nodes and worsen survival. A simplified two-tier system labels pattern A and some B tumors as low risk, and pattern C and B tumors with vessel invasion as high risk. What has been missing is an understanding of what is happening at the molecular level inside these different patterns, especially in the living neighborhood of the tumor—the nearby supporting tissue and immune cells that can either restrain or encourage cancer growth.
Reading gene activity in place
To tackle this, the researchers used a technology called spatial transcriptomics on seven surgically removed tumors that contained more than one invasion pattern in the same specimen. This clever design let them compare low-risk and high-risk areas within a single patient, reducing background differences from person to person. With the GeoMX platform, they selected dozens of small regions that included both cancer cells and the surrounding stromal immune microenvironment (SIME). Fluorescent markers were used to separate RNA coming from tumor epithelium versus the nearby non-tumor tissue. They then sequenced the RNA to see which genes were turned on or off in each compartment and used statistical tools to find consistent changes tied to high-risk invasion.
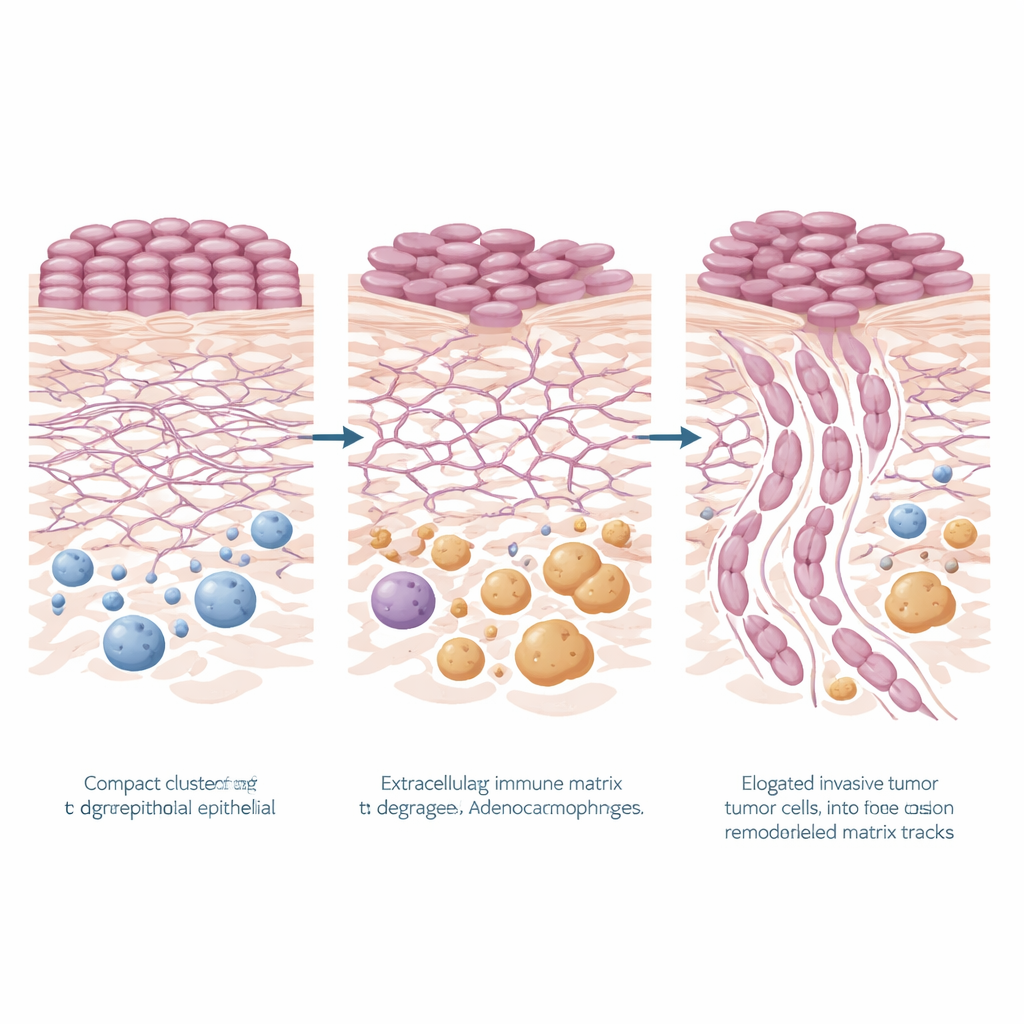
How the tumor reshapes its scaffolding
A striking pattern emerged: genes involved in remodeling the body’s structural scaffolding, known as the extracellular matrix, were strongly increased in high-risk regions, both in the tumor cells and in the surrounding tissue. Pathways linked to matrix breakdown, matrix organization, cell attachment, and related signaling (including PI3K–Akt signaling) were all elevated. Several key genes stood out in the cancer epithelium—KRT6A, TNC, LAMC2, and FN1—many of which encode proteins that help cells attach, move, or reshape their environment. In the nearby stroma, genes such as MMP9 and POSTN, associated with cutting and rebuilding matrix fibers and with more aggressive tumor behavior, were also increased. Together, these changes paint a picture of high-risk tumors that are actively carving new paths through tissue and building a microenvironment that favors invasion.
Immune cells that help rather than hinder
The surrounding immune landscape also shifted in more dangerous patterns. Gene signatures in the SIME pointed to heightened activity of the innate arm of the immune system and increased presence of macrophages, a type of white blood cell. Using computational methods, the team inferred that so‑called M2-like macrophages—often associated with wound healing and tumor support rather than attack—were more abundant in high-risk regions. This was backed up at the protein level: tissue staining for CD68, a marker of macrophages, showed denser macrophage populations around the most invasive tumor patterns. The data suggest that remodeled matrix and macrophage-rich stroma may work together to create a nurturing niche that helps the cancer push deeper.
A simple gene score that signals trouble
To explore clinical impact, the authors built a four-gene signature from those strongly upregulated in high-risk tumor epithelium and more expressed in tumor than normal cervix: KRT6A, TNC, LAMC2, and FN1. They combined the expression of these genes into a single score and tested it in an independent set of cervical adenocarcinomas from The Cancer Genome Atlas. Even in this small group, tumors with higher scores tended to have worse overall survival, and a cut-off value could separate patients into lower- and higher-risk groups better than stage alone. While the numbers are modest and need validation in larger cohorts, this kind of gene-based tool resembles tests already used in breast cancer to guide treatment decisions.
What it all means going forward
In accessible terms, this study shows that more dangerous HPV-associated endocervical adenocarcinomas are not just “bigger” versions of safer tumors; they are biologically different. High-risk regions are marked by cancer cells and neighboring tissue that are jointly remodeling the tissue scaffold and recruiting helper immune cells, especially certain macrophages, to support invasion. The four-gene signature distilled from these changes hints at a future where simple molecular tests could flag patients whose tumors are likely to behave aggressively, even when stage looks early. The findings also point to new treatment angles: drugs that target extracellular matrix remodeling or reshape the innate immune response may hold promise for women with high-risk invasion patterns in this type of cervical cancer.
Citation: Axelrod, M.L., Zhou, R. & Sun, L. Spatial transcriptomic landscape of invasion patterns in human papillomavirus-associated endocervical adenocarcinoma. Sci Rep 16, 13246 (2026). https://doi.org/10.1038/s41598-026-43717-z
Keywords: cervical adenocarcinoma, HPV-related cancer, tumor microenvironment, extracellular matrix remodeling, prognostic gene signature