Clear Sky Science · en
Cerium driven active site relocation in spinel Co3O4 enables stable chlorine evolution in acidic media
Why making chlorine more sustainable matters
Chlorine is hidden in the background of daily life—from safe drinking water to plastics and medicines—but making it consumes enormous amounts of electricity and relies on scarce, costly precious metals. This study tackles a long-standing problem: how to build a cheaper, longer‑lasting anode material for chlorine production that can survive the harsh acidic, salty environment inside industrial chlor‑alkali plants without quickly corroding or falling apart.
Moving beyond expensive precious metals
Today’s chlorine plants mostly use anodes made from oxides of ruthenium and iridium. These noble metals are highly active and resistant to corrosion, but they are rare and expensive, and they also tend to catalyze unwanted oxygen formation, which wastes energy and reduces chlorine output. More common metal oxides based on 3d metals such as cobalt are attractive because they are cheaper and tunable, yet they usually dissolve or restructure rapidly in acidic, chloride-rich solutions. The main weakness is that, on these materials, oxygen atoms in the crystal lattice often act as the reactive sites. Those oxygen sites are good at grabbing chloride ions, but they are also easily “etched away,” undermining the structure.
Redesigning where the reaction happens
The authors propose a different strategy: instead of letting oxygen atoms do the work, deliberately shift the key reaction sites onto metal atoms that are more robust. They achieve this by inserting single cerium atoms into specific positions of a cobalt oxide known as spinel Co3O4 and shaping it into a three‑dimensional ordered macroporous network. Careful structural and spectroscopic measurements show that cerium atoms selectively occupy octahedral cobalt positions in the lattice and subtly distort the surrounding cobalt–oxygen building blocks. This distortion creates “unsaturated” cobalt centers near the surface that are primed to bind chloride ions directly, while keeping the overall spinel framework intact. The porous, ordered structure at the micron scale increases surface area and helps move reactants and gas bubbles efficiently at high current.

Proving that cerium reroutes the reaction
To find out whether the reaction really moves from oxygen sites to cobalt sites, the team used a suite of in situ techniques—probing the catalyst while it is working. Raman spectroscopy detected the formation of cobalt–chlorine bonds on the cerium‑doped material under reaction conditions, but not on undoped Co3O4. Infrared measurements with oxygen isotopic labeling revealed chlorine bound to lattice oxygen on the undoped catalyst, signaling an oxygen‑centered pathway, while on the cerium‑doped version such oxygen–chlorine signals were only transient before cobalt–chlorine interactions took over. Mass spectrometry confirmed that the doped catalyst produced almost exclusively chlorine gas with hardly any oxygen, even at high currents. Together, these observations show that cerium reshapes the local environment so that chloride prefers to bind to cobalt rather than to oxygen, reducing the corrosive participation of lattice oxygen.
How atomic-scale tuning boosts performance
Density functional theory calculations helped explain these experimental findings. On pristine Co3O4, the most favorable place for chloride adsorption is a bridging oxygen atom between cobalt centers; attempts to place chloride on cobalt relaxed back to nearby oxygen, matching the observed oxygen‑centered mechanism. After substituting a cobalt site with cerium, the surrounding polyhedra open up, creating a coordinatively unsaturated cobalt site whose electronic levels are shifted so that chloride binds with just the right strength: strong enough to react efficiently, but not so strong that products cannot leave. The same calculations show that pathways leading to oxygen evolution become energetically less favorable, which accounts for the near‑unity chlorine selectivity. In essence, cerium co‑tunes geometry and electron distribution to favor a cobalt‑centered chlorine pathway while suppressing routes that damage the lattice.
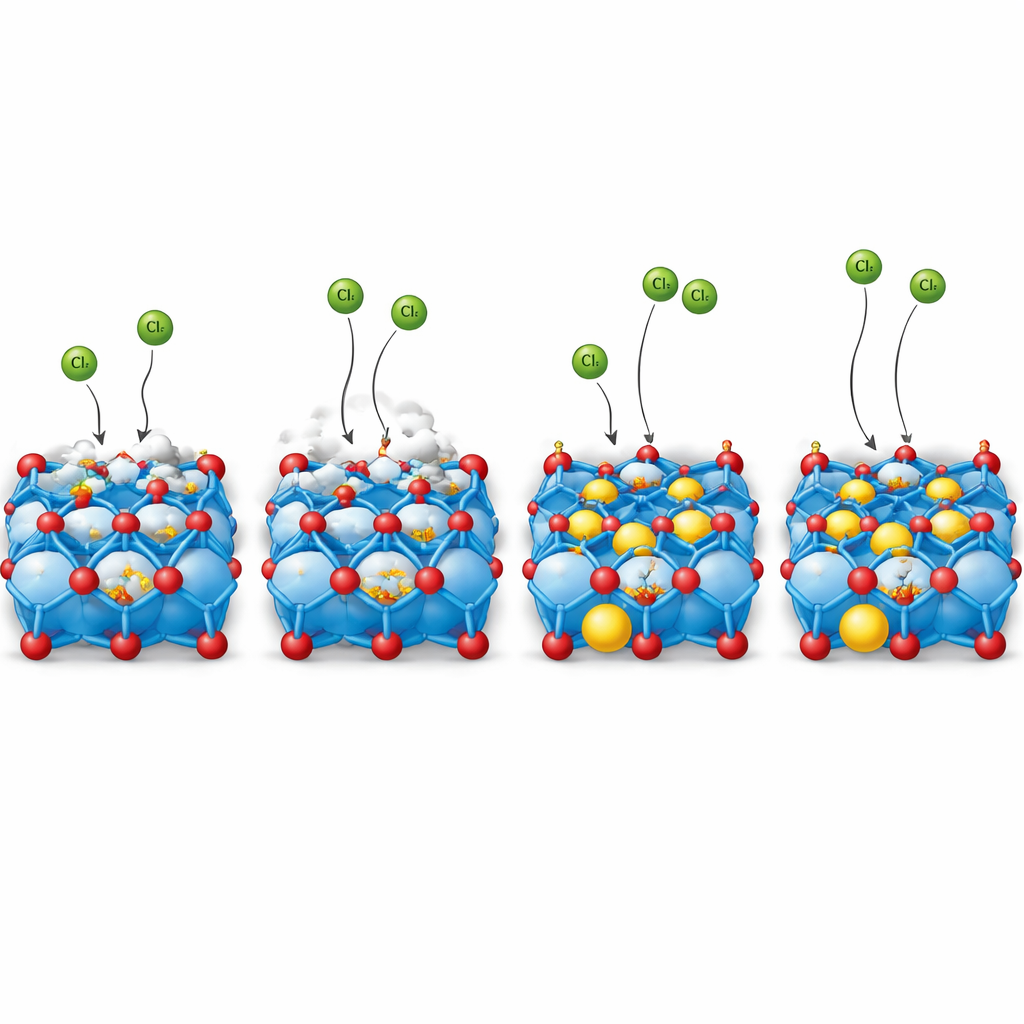
From lab tests to industrial-like operation
Electrochemical tests in concentrated sodium chloride at low pH show that the cerium‑doped porous Co3O4 catalyst reaches industrially relevant current densities with remarkably low extra voltage and with about 99% of the current going to chlorine rather than oxygen. In a flow cell, it runs stably for hundreds of hours with only minor cobalt loss, far outlasting undoped Co3O4. When assembled into a practical chlor‑alkali cell with brine on one side and caustic solution on the other, the new anode delivers high currents at lower cell voltages than both conventional cobalt oxide and a commercial precious‑metal anode, and maintains performance for more than 500 hours, even at kiloampere‑per‑square‑meter current densities typical of industry.
What this means for future chlorine production
For a non‑specialist, the key message is that the authors have shown how to “move” the business end of a reaction from fragile oxygen atoms to sturdier metal atoms inside a common oxide, simply by sprinkling in a tiny amount of cerium at the right lattice sites and giving the material an open, sponge‑like structure. This shift makes the catalyst both more efficient and far more durable in an extremely aggressive environment, offering a blueprint for chlorine production that relies less on scarce noble metals. More broadly, the concept of relocating active sites away from vulnerable atoms could guide the design of longer‑lived, high‑current electrocatalysts for many other large‑scale chemical processes.
Citation: Mao, Z., Zhang, J., Tu, T. et al. Cerium driven active site relocation in spinel Co3O4 enables stable chlorine evolution in acidic media. Nat Commun 17, 3763 (2026). https://doi.org/10.1038/s41467-026-70443-x
Keywords: chlorine evolution reaction, chlor-alkali electrolysis, cobalt oxide catalyst, cerium doping, electrocatalysis stability