Clear Sky Science · en
SPT6 maintains epidermal homeostasis by inhibiting an NF-κB-positive feedback loop to prevent excessive inflammation
Why calming the skin’s alarm system matters
The skin is our largest organ and our first shield against the outside world. To protect us, skin cells must detect danger quickly, yet avoid setting off needless alarms that lead to chronic redness, flaking, and pain. This study uncovers how a little-known protein called SPT6 helps keep the skin’s alarm system under control. When SPT6 is lost in key skin cells, the tissue tips into psoriasis‑like inflammation and heals more slowly after injury, revealing how fragile the balance between protection and overreaction can be.
How skin cells act as front‑line sentries
The outer layer of the skin, the epidermis, is built from keratinocytes—cells that constantly renew themselves and form a tight barrier. These cells do far more than sit passively; they sense germs and damage and release chemical signals that summon immune cells. The authors asked whether special stem‑like cells at the base of the epidermis not only raise alarms, but also actively dampen excessive inflammation. They focused on SPT6, a protein that helps genes switch on properly, to see if it acts as a built‑in brake on inflammatory signals.
When a gatekeeper is removed from mouse skin
Using genetically engineered mice, the team selectively deleted the Supt6 gene (which produces SPT6) in basal keratinocytes. Within days, the animals developed thickened, scaly skin on the back, ears, and paws, along with hair loss and weight decline. Microscopy and ultrastructure studies showed hallmark features seen in psoriasis: an overly thick epidermis, sticky layers of dead cells on the surface, faulty cell‑cell attachments, and immune cells crowding into the tissue. Despite these dramatic skin changes, other K14‑positive epithelial sites such as the tongue and esophagus remained structurally normal, pointing to a skin‑focused role for SPT6.
Inflamed yet over‑mature skin and slower repair
Gene‑activity profiling revealed that thousands of genes shifted when SPT6 was lost. Cell‑adhesion and Wnt repair pathways were dialed down, while genes controlling cell division, skin maturation, and inflammatory responses were strongly boosted. Markers of late epidermal differentiation were elevated, meaning the skin appeared overly “mature” yet disordered. When the researchers created full‑thickness wounds, SPT6‑deficient mice healed significantly more slowly. Many genes involved in the Wnt pathway—known to drive hair growth and wound closure—were reduced, and prior binding data showed SPT6 sits on several of these repair genes. This suggests SPT6 supports healthy regeneration while keeping inflammatory programs in check.
A psoriasis‑like signature that arises from within
Inflammation‑related genes such as Il1b, Il6, Tnf, and S100a8/a9 were sharply increased in SPT6‑deficient skin. The pattern of gene changes strongly resembled that seen in a standard mouse model of psoriasis and in human psoriatic lesions. Neutrophils accumulated on the skin surface in structures reminiscent of Munro’s microabscesses, a classic psoriasis feature. Yet eliminating bacteria with broad‑spectrum antibiotics, screening for fungi and common skin viruses, and even transferring skin microbes between mice did not alter the disease. Single‑cell RNA sequencing showed that specific basal and intermediate keratinocyte subgroups expanded and adopted highly inflammatory and pro‑differentiation programs, indicating that the drive toward disease comes from the keratinocytes themselves rather than from invading microbes.
How SPT6 blocks a self‑amplifying inflammatory loop
Zooming in on mechanism, the researchers studied human keratinocytes in culture. When they reduced SPT6 and then mimicked viral or damage signals, the cells unleashed a wave of inflammatory genes controlled by NF‑κB, a master switch for immune responses. A drug that blocks NF‑κB partly reversed this surge and eased skin inflammation in SPT6‑deficient mice. The team then discovered that SPT6 binds an enhancer region near the RELA gene, which encodes the key NF‑κB subunit p65. Without SPT6, more p65 bound both this enhancer and the RELA promoter, creating a positive feedback loop: p65 drives more of its own gene’s activity, which in turn further boosts inflammatory gene expression.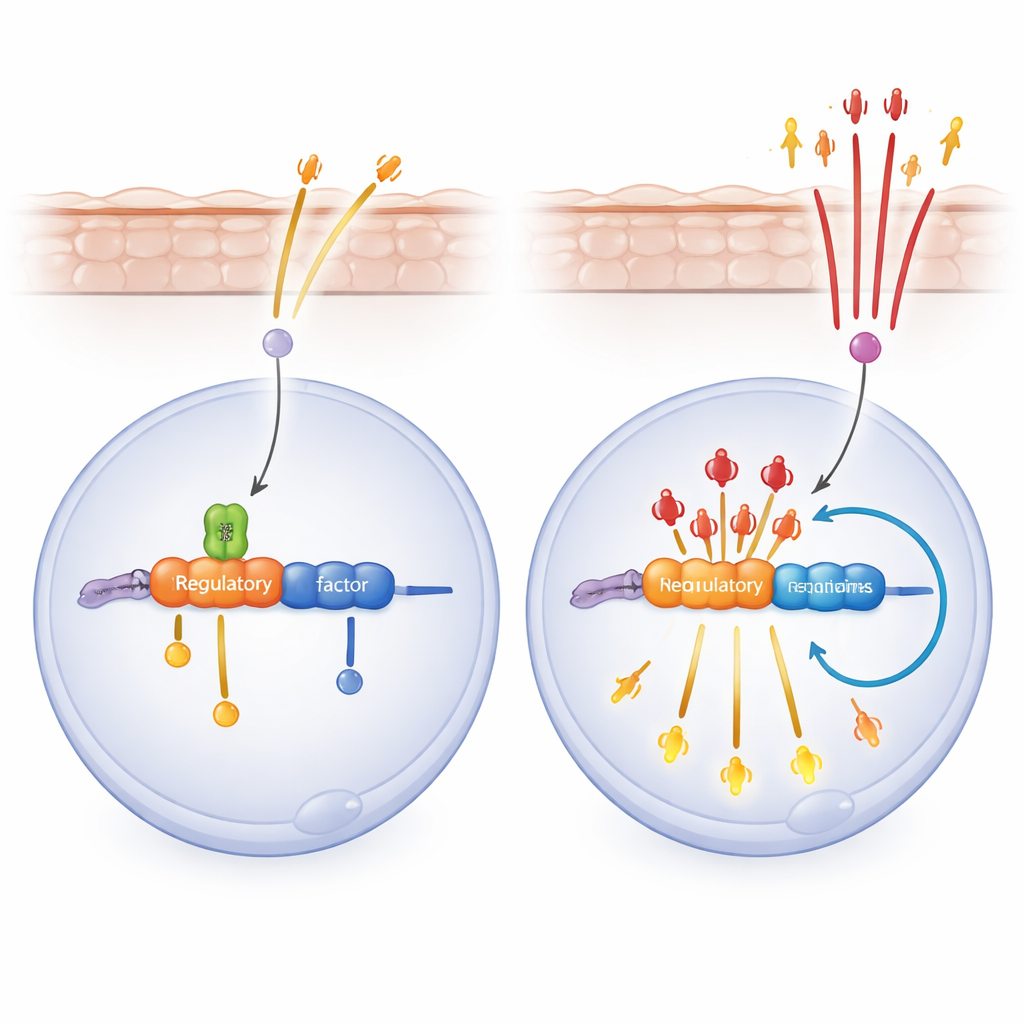
What this means for understanding and treating skin disease
To a lay reader, the message is that the skin is naturally poised to ignite inflammation, but it stays quiet only because internal brakes like SPT6 constantly hold the response in check. When SPT6 is removed from basal keratinocytes, these cells over‑differentiate, send stronger distress signals, and drag the tissue into psoriasis‑like disease and poor wound repair, all without an obvious infection. By showing that SPT6 restrains an NF‑κB feedback loop at the RELA gene, this work highlights a precise control point that could be targeted to calm chronic skin inflammation while preserving the body’s ability to fight real threats.
Citation: Sun, Y., Xu, S., Wang, D. et al. SPT6 maintains epidermal homeostasis by inhibiting an NF-κB-positive feedback loop to prevent excessive inflammation. Cell Mol Immunol 23, 471–490 (2026). https://doi.org/10.1038/s41423-026-01410-1
Keywords: skin inflammation, psoriasis, keratinocytes, NF-kappaB signaling, wound healing