Clear Sky Science · de
De-novo-Erzeugung kovalenter Wirkstoffe mit verbesserter Wirkstoffähnlichkeit und Sicherheit
Warum intelligentere kovalente Wirkstoffe wichtig sind
Krebstherapien und antivirale Medikamente wirken oft, indem sie sich an Proteine in unseren Zellen anlagern. Eine besonders potente Wirkstoffklasse, die kovalenten Wirkstoffe, geht einen Schritt weiter: Sie bilden enge, langanhaltende Bindungen mit ihren Zielmolekülen, was sehr wirkungsvoll sein kann. Dieselbe «Haftung» kann jedoch auch schwere Nebenwirkungen verursachen, wenn die Wirkstoffe an falsche Proteine binden. Diese Studie stellt CovaGEN vor, ein neues KI‑System, das kovalente Wirkstoffkandidaten von Grund auf entwirft und dabei sowohl Wirksamkeit als auch Sicherheit berücksichtigt.
Vom Trial‑and‑Error zur klugen Gestaltung
Die traditionelle Entdeckung kovalenter Wirkstoffe ist langsam und teuer. Chemiker beginnen entweder mit einem bekannten Wirkstoff und ergänzen ein reaktives «Griffstück», oder sie starten mit einem reaktiven Fragment und passen den Rest des Moleküls mühsam an. Computerunterstützte Methoden helfen, indem sie große Bibliotheken bekannter Verbindungen durchsuchen, sind dabei aber auf bereits Existierendes beschränkt. Jüngste Fortschritte im Deep Learning können ganz neue Moleküle erzeugen, doch diese Werkzeuge konzentrieren sich meist auf konventionelle, nicht‑kovalente Wirkstoffe und vernachlässigen oft entscheidende Eigenschaften wie die Synthesierbarkeit, die Wirkstoffähnlichkeit oder mögliche Toxizität. CovaGEN will all diese Probleme gleichzeitig angehen.
Eine verborgene Karte der wirkstoffähnlichen Chemie
Die Autoren bauten zunächst eine «Karte» des chemischen Raums mithilfe von mehr als einer Million wirkstoffähnlicher Moleküle aus der öffentlichen ZINC‑Datenbank. Sie trainierten einen Typ von neuronalen Netzwerken, einen variationalen Autoencoder, um jedes Molekül in einen kompakten numerischen Code bzw. einen latenten Vektor zu komprimieren und anschließend zu rekonstruieren. Dieser Schritt lehrt das Modell die grundlegende Grammatik der Arzneimittelchemie: Was realistisch wirkt, welche Regeln für orale Medikamente zu beachten sind und was synthetisch praktikabel ist. Auf dieser gelernten Karte wurde ein zweites Modell auf Basis von Diffusion — ein Prozess, der Struktur schrittweise in Rauschen verwandelt und lernt, diesen Prozess umzukehren — trainiert, um in diesem Raum zu wandern und neue latente Vektoren zu erzeugen, die in gültige, vielfältige und realistische Moleküle dekodieren.
Moleküle auf Proteine und reaktive Haken abstimmen
Als Nächstes lernt CovaGEN, Moleküle für spezifische Proteinzielstrukturen zu entwerfen. Das System nimmt die Aminosäuresequenz eines Proteins und kodiert sie mit einem leistungsfähigen Sprachmodell für Proteine, das Muster in Bezug auf Struktur und Funktion erfasst. Während der Diffusion richtet das Modell seine Aufmerksamkeit kreuzweise auf diese Proteindarstellung, sodass die erzeugten Moleküle eher dazu neigen, in die Bindungsstelle des Ziels zu passen. In Tests an einem umfangreichen Benchmark‑Datensatz erreichten diese zugeschnittenen Moleküle nicht nur gute Werte in Bezug auf vorhergesagte Bindungsstärke, sondern zeigten auch bessere Wirkstoffähnlichkeit und synthetische Zugänglichkeit als Moleküle aus mehreren hochmodernen 3D‑Design‑Werkzeugen. Um diese nicht‑kovalenten Binder in kovalente Kandidaten zu verwandeln, ergänzte das Team einen Klassifikator, der den Diffusionsprozess in Richtung Moleküle mit bestimmten reaktiven Gruppen — sogenannten kovalenten Warheads — lenkt, ohne die Gesamtqualität zu opfern.
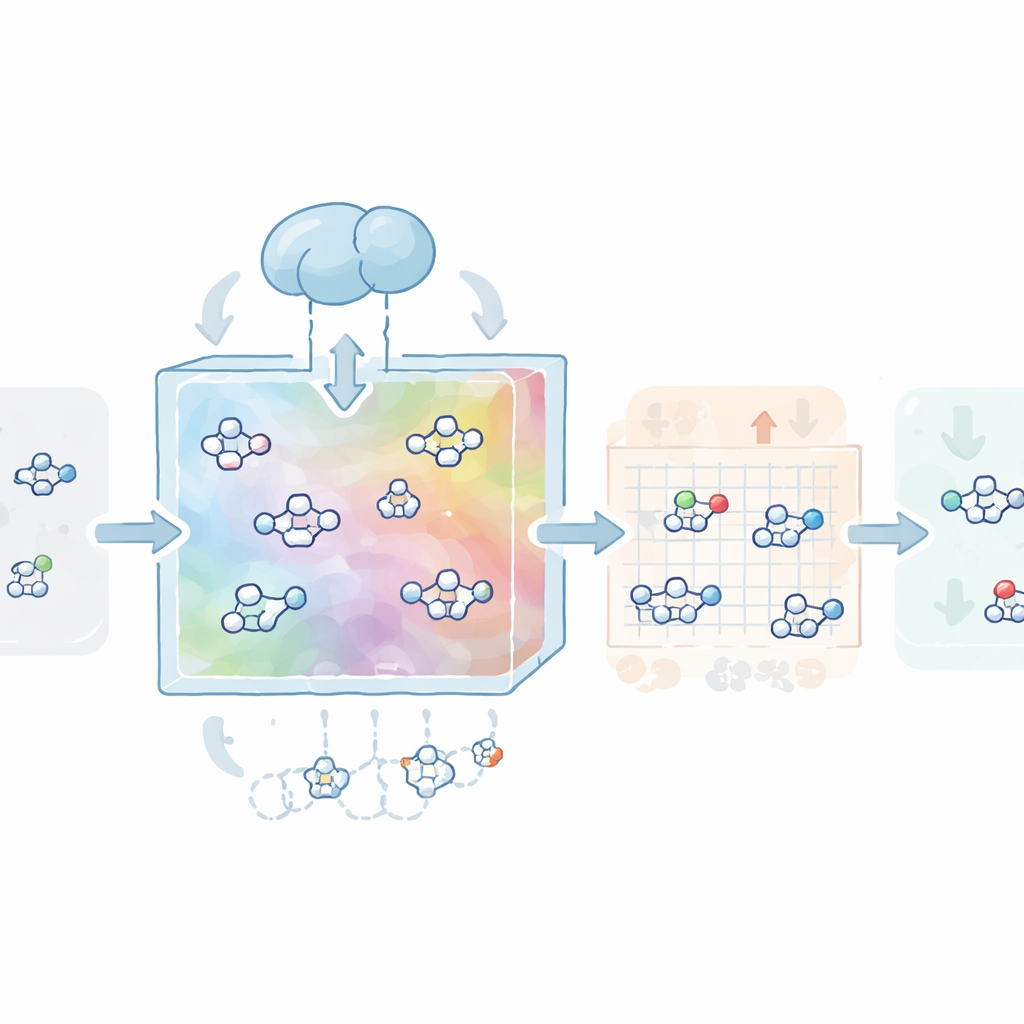
Sicherheit von Anfang an einbauen
Da kovalente Wirkstoffe bleibenden Schaden anrichten können, wenn sie falsche Ziele treffen, ist Sicherheit ein zentrales Anliegen. Die Forscher behandelten Toxizität daher als zu optimierende Größe und nicht als nachträglichen Gedankengang. Sie trainierten prädiktive Modelle für akute Toxizität und organspezifische Risiken und setzten anschließend Reinforcement Learning ein, um das Diffusionsmodell so nachzujustieren, dass es Moleküle mit geringerer vorhergesagter Toxizität bevorzugt. Nach diesem Training erzeugte CovaGEN Verbindungen, die gute Bindungs‑ und wirkstoffähnliche Merkmale beibehielten, aber weniger wahrscheinlich strukturelle «Rote Flaggen» aufwiesen, die mit schädlichen Effekten assoziiert sind. Wichtig ist, dass die Methode dies erreichte, ohne in einen engen Satz ähnlicher Moleküle zu kollabieren, sodass die chemische Vielfalt erhalten blieb.
Die Methode auf die Probe stellen
Um das Potenzial in der Praxis zu zeigen, bat das Team CovaGEN, kovalente Inhibitoren für zwei besonders relevante Ziele zu entwerfen: eine mutierte Form des EGFR‑Proteins, das bei medikamentenresistentem Lungenkrebs eine Rolle spielt, und die Hauptprotease von SARS‑CoV‑2, dem Virus hinter COVID‑19. Für jedes Protein generierte das System zahlreiche Kandidatenmoleküle mit den geeigneten Warheads. Rechnerische Docking‑Studien deuteten darauf hin, dass CovaGENs Entwürfe eher Posen einnehmen, die kovalente Bindungen ermöglichen, als Verbindungen, bei denen Warheads einfach an nicht‑kovalente Designs angefügt wurden. Mehrere Top‑Kandidaten erreichten zudem in kombinierten Bewertungen von vorhergesagter Bindung, Wirkstoffähnlichkeit und synthetischer Einfachheit Referenzmedikamente oder übertrafen sie.
Was das für zukünftige Arzneimittel bedeutet
CovaGEN zeigt, dass moderne generative KI mehr kann, als nur Moleküle zu erfinden, die fest an Proteine binden. Indem sie in einem Raum von grundsätzlich wirkstoffähnlichen Strukturen arbeitet, die Hinzufügung reaktiver Gruppen führt und explizit von Toxizität wegsteuert, bringt das System die Entdeckung kovalenter Wirkstoffe in Richtung eines ganzheitlicheren und automatisierten Prozesses. Obwohl weitere Laboruntersuchungen nötig sind, um zu bestätigen, wie sich diese virtuellen Moleküle in biologischen Systemen verhalten, eröffnet der Ansatz einen Weg zu schnellerer und breiterer Erkundung kovalenter Arzneimittel, die nicht nur potent, sondern auch sicherer für Patientinnen und Patienten sind.
Zitation: Zhang, W., Liu, T., Dong, X. et al. De novo covalent drug generation with enhanced drug-likeness and safety. Commun Biol 9, 446 (2026). https://doi.org/10.1038/s42003-026-09725-5
Schlüsselwörter: kovalente Wirkstoffe, generative KI, Wirkstoffforschung, Diffusionsmodelle, Toxizitätsvorhersage