Clear Sky Science · de
Vergleichende Studie von Armchair- und Zigzag-Graphen-Quantenpunkten als HIV-1-Protease-Inhibitoren
Ein winziges Kohlenstoffwerkzeug im Kampf gegen HIV
Medikamente, die verhindern, dass HIV Kopien von sich selbst herstellt, haben eine tödliche Infektion in eine behandelbare Krankheit verwandelt, doch das Virus kann weiterhin Resistenzen gegen bestehende Wirkstoffe entwickeln. Diese Studie untersucht einen neuen Typ von Wirkstoffkandidaten aus Graphen-Quantenpunkten – ultrakleine Kohlenstoffflocken im Nanometerbereich – um zu prüfen, ob sie an ein zentrales virales Enzym, die HIV-1-Protease, binden und es potenziell abschalten könnten.
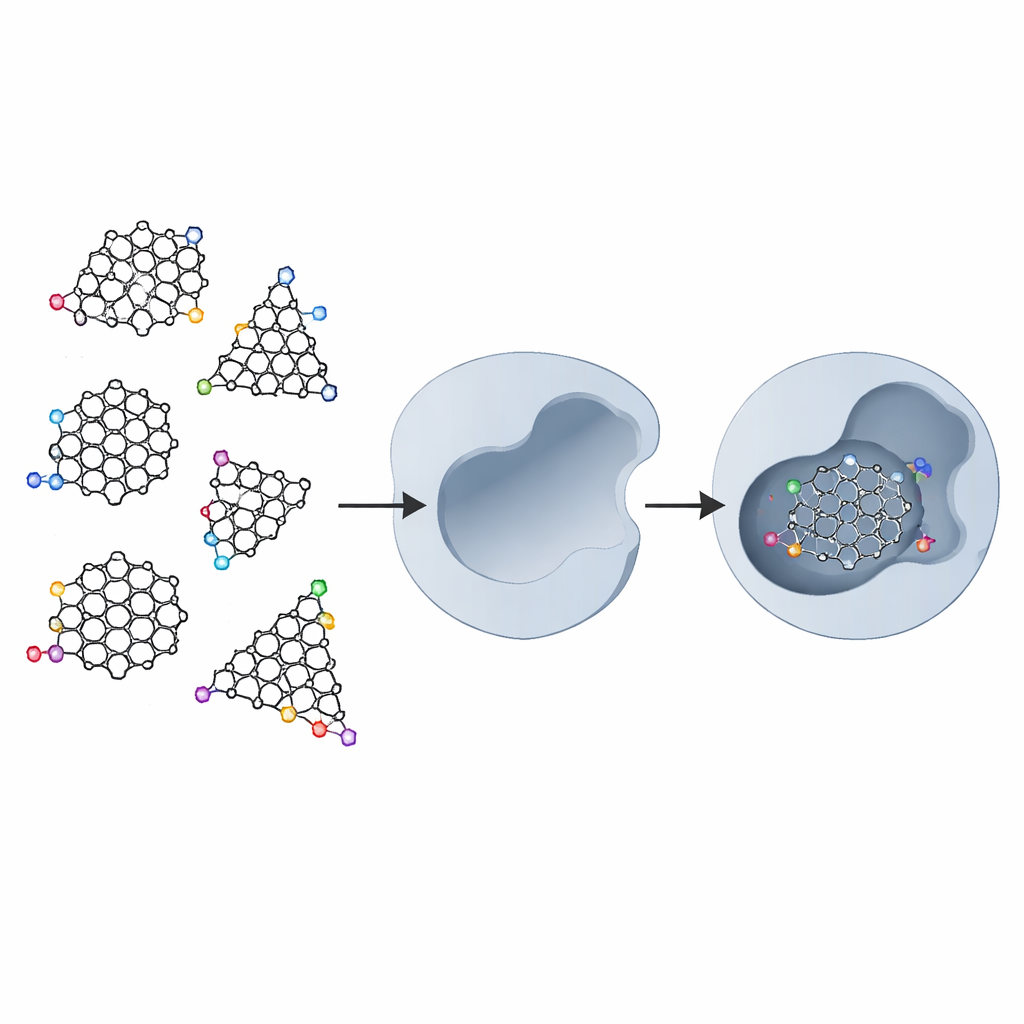
Was diese Kohlenstoffdots besonders macht
Graphen ist eine Lage von Kohlenstoffatomen, die wie Hühnerdraht angeordnet sind. Wenn diese Lage in sehr kleine Stücke zerschnitten wird, sogenannte Graphen-Quantenpunkte, zeigen sie neue Eigenschaften, weil Elektronen in einem so winzigen Raum eingeschlossen werden und weil die Kanten der Stücke chemisch aktiv werden. Die Punkte können dreieckig oder sechseckig geschnitten sein, und ihre Ränder können unterschiedliche Muster aufweisen – "armchair"- oder "zigzag"-Anordnungen der Atome. Darüber hinaus können Chemiker zusätzliche chemische Gruppen an den Kanten anbringen, um die Wasserlöslichkeit zu verbessern und wie die Dots mit biologischen Molekülen interagieren. Diese kombinierten Eigenschaften machen Graphen-Quantenpunkte nicht nur für Elektronik und Bildgebung, sondern auch für die Medizin vielversprechend.
Entwurf kohlenstoffbasierter Proteasehemmer
Die Forscher untersuchten fünf Grundtypen von Graphen-Quantenpunkten: eine unmodifizierte Fläche, dreieckige und sechseckige Stücke mit Armchair-Kanten sowie dreieckige und sechseckige Stücke mit Zigzag-Kanten. Anschließend "dekorierten" sie jedes dieser Modelle mit einer kleinen Ringsystemeinheit (Pyrrolidin), einem Benzolring und schließlich zwei Hydroxymethylcarbonyl-(HMC)-Gruppen. Die HMC-Gruppen wurden ausgewählt, weil sie Wasserstoffbrücken mit zwei Asparaginsäure-Resten ausbilden können, die im Zentrum der aktiven Stelle der HIV-1-Protease liegen. In der realen Enzymumgebung sind nahezu alle benachbarten Aminosäuren hydrophob, doch diese beiden Asparaginsäuren sind hydrophil und bilden so einen natürlichen Andockpunkt für gezielt platzierte chemische Haken.
Reaktivität am Computer testen
Statt im Nasslabor arbeitete das Team mit hochrangigen quantenchemischen Rechnungen, um vorherzusagen, wie sich diese Designer-Dots verhalten. Sie optimierten jede Struktur und berechneten Größen, die auf chemische Reaktivität hinweisen, wie das totale Dipolmoment (ein Maß dafür, wie ungleichmäßig die Ladung verteilt ist) und die Energiedifferenz zwischen dem höchsten besetzten und dem niedrigsten unbesetzten Elektronenzustand. Ein großes Dipolmoment kombiniert mit einer kleinen Lücke deutet gewöhnlich darauf hin, dass ein Molekül eher zu Wechselwirkungen neigt. Unter allen Entwürfen stach ein dreieckiger Dot mit Zigzag-Kante und angefügten Pyrrolidin- und HMC-Gruppen hervor: Er zeigte die höchste Polarität und die kleinste Lücke. Die Wissenschaftler untersuchten zudem, wie sich die Elektronenwolke über jeden Dot verteilt und wie viele elektronische Zustände verfügbar sind, was weiter bestätigt, dass bestimmte Formen und Kantenmuster das Material reaktionsfreudiger machen.
Wie gut die Dots das Enzym greifen
Um zu verstehen, ob diese Quantenpunkte das HIV-1-Protease-Enzym wirklich umfassen könnten, simulierten die Forscher ihre Wechselwirkung mit zwei Asparaginsäure-Resten, die die aktive Stelle des Enzyms nachahmen. Mithilfe der sogenannten Quantum Theory of Atoms in Molecules untersuchten sie die feinen Details der Elektronendichte an den Stellen, wo Bindungen entstehen könnten. Alle modifizierten Dots bildeten stabile Komplexe, doch das zigzag-dreieckige Design mit HMC-Gruppen zeigte besonders starke Wechselwirkungen, wobei einige Kontakte einen teilweise kovalenten Charakter annahmen – näher an einer echten chemischen Bindung als an einer flüchtigen Anziehung. Ein weiterer wichtiger Faktor war die Größe: Die am besten passende Struktur, basierend auf einer unveränderten Fläche modifiziert mit Pyrrolidin, Benzol und zwei HMC-Gruppen, maß etwa 9,3 Å im Durchmesser und passte damit gut in die ungefähr 10 Å breite Kavität der realen Protease-Aktivenstelle.
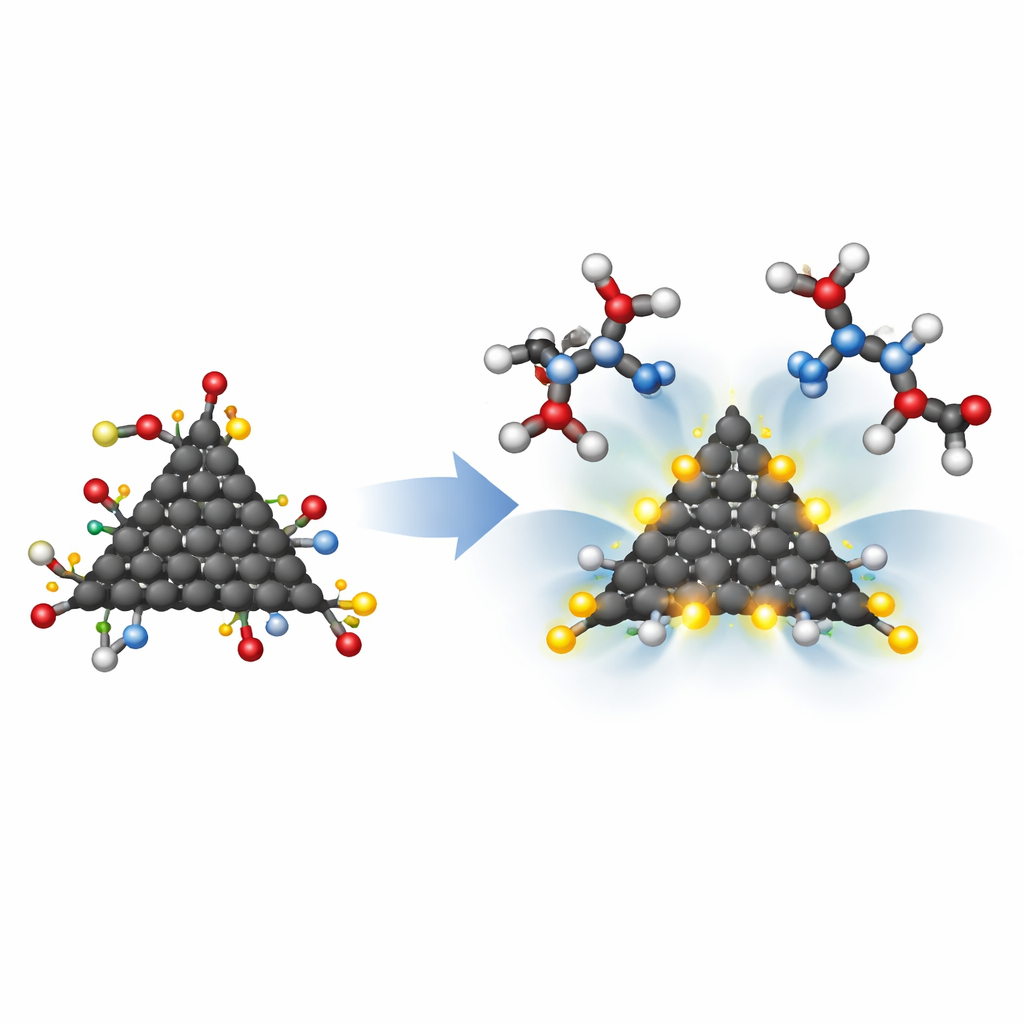
Warum das für zukünftige HIV-Behandlungen wichtig ist
Durch die Kombination von intelligenter Nanoarchitektur mit detaillierten Computermodellen zeigt diese Arbeit, dass winzige Kohlenstoffdots so angepasst werden können, dass sie sowohl in die Tasche der HIV-1-Protease passen als auch deren zentrale Asparaginsäurereste fest erfassen. Die vielversprechendsten Varianten sind klein genug, um in die Enzymkavität einzudringen, polar und reaktiv genug, um starke Wasserstoffbrücken zu bilden, und elektronisch stabil, sobald sie gebunden sind. Obwohl es sich um frühe, theoretische Ergebnisse und nicht um fertige Medikamente handelt, legen sie dar, wie Form, Kantenmuster und angefügte chemische Gruppen von Graphen-Quantenpunkten gemeinsam deren Fähigkeit steuern, als HIV-1-Protease-Hemmer zu wirken. Diese Roadmap könnte künftig das Design einer neuen Klasse kohlenstoffbasierter antiviraler Materialien leiten.
Zitation: Ibrahim, A., Elhaes, H. & Ibrahim, M.A. Comparative study of armchair and zigzag graphene quantum dots as HIV-1 protease inhibitors. Sci Rep 16, 14650 (2026). https://doi.org/10.1038/s41598-026-48709-7
Schlüsselwörter: Graphen-Quantenpunkte, HIV-1-Protease, Nanomedizin, computerbasiertes Wirkstoffdesign, Kohlenstoff-Nanomaterialien