Clear Sky Science · de
Neue Indol-gekoppelte 1,2,4‑Triazol-Derivate als duale FAK-Inhibitoren und Apoptoseinduktoren, die Überleben und Migration bei triple-negativem Brustkrebs in vitro angreifen
Warum diese Forschung für Patientinnen und Patienten wichtig ist
Triple‑negativer Brustkrebs gehört zu den schwierigsten Formen von Brustkrebs, weil ihm die Hormonrezeptoren fehlen, auf die viele moderne Medikamente abzielen. Betroffene sehen sich häufig einer aggressiven Erkrankung, begrenzten Therapieoptionen und belastenden Chemotherapien gegenüber. Diese Studie verfolgt einen anderen Ansatz: eine neue Familie niedermolekularer Verbindungen, die einen zentralen Überlebensschalter in Krebszellen ausschalten und diese zur Selbstzerstörung treiben sollen, dabei aber gesunde Zellen möglichst verschonen.
Ein neuer Weg, einen aggressiven Krebs anzugreifen
Die Forschenden konzentrierten sich auf triple‑negativen Brustkrebs, der dazu neigt, früh zu streuen und Standardtherapien zu widerstehen. Diese Tumoren sind häufig auf ein Protein angewiesen, das als Focal‑Adhesion‑Kinase (FAK) bezeichnet wird und Krebszellen beim Anhaften, Bewegen und Überleben unter Stress unterstützt. Ist FAK überaktiv, werden Tumoren invasiver und schwerer zu eliminieren. Da es bislang kaum breit eingesetzte zielgerichtete Medikamente für diese Krebsform gibt, ist die Blockade von FAK eine attraktive Strategie, um Tumorwachstum zu bremsen und die Ausbreitung zu verhindern.
Maßgeschneiderte Krebshemmer entwerfen
Um FAK anzugreifen, synthetisierte das Team eine Reihe neuer Moleküle aus zwei gut bekannten Bausteinen der Chemie: Indolen und 1,2,4‑Triazolen. Durch unterschiedliche Verknüpfungen und Anordnungen dieser Elemente entstanden mehrere verwandte Verbindungen, die sie gegen zwei Brustkrebszelllinien und normale menschliche Fibroblasten testeten. Drei Kandidaten, bezeichnet als 3c, 4c und 5c, zeichneten sich dadurch aus, dass sie das Wachstum von Krebszellen bei vergleichsweise niedrigen Dosen verlangsamten und zugleich weniger schädlich für normale Zellen waren. Unter ihnen erwies sich Verbindung 4c, die zwei Indolringe über einen Triazolkern verbindet, als besonders vielversprechend. 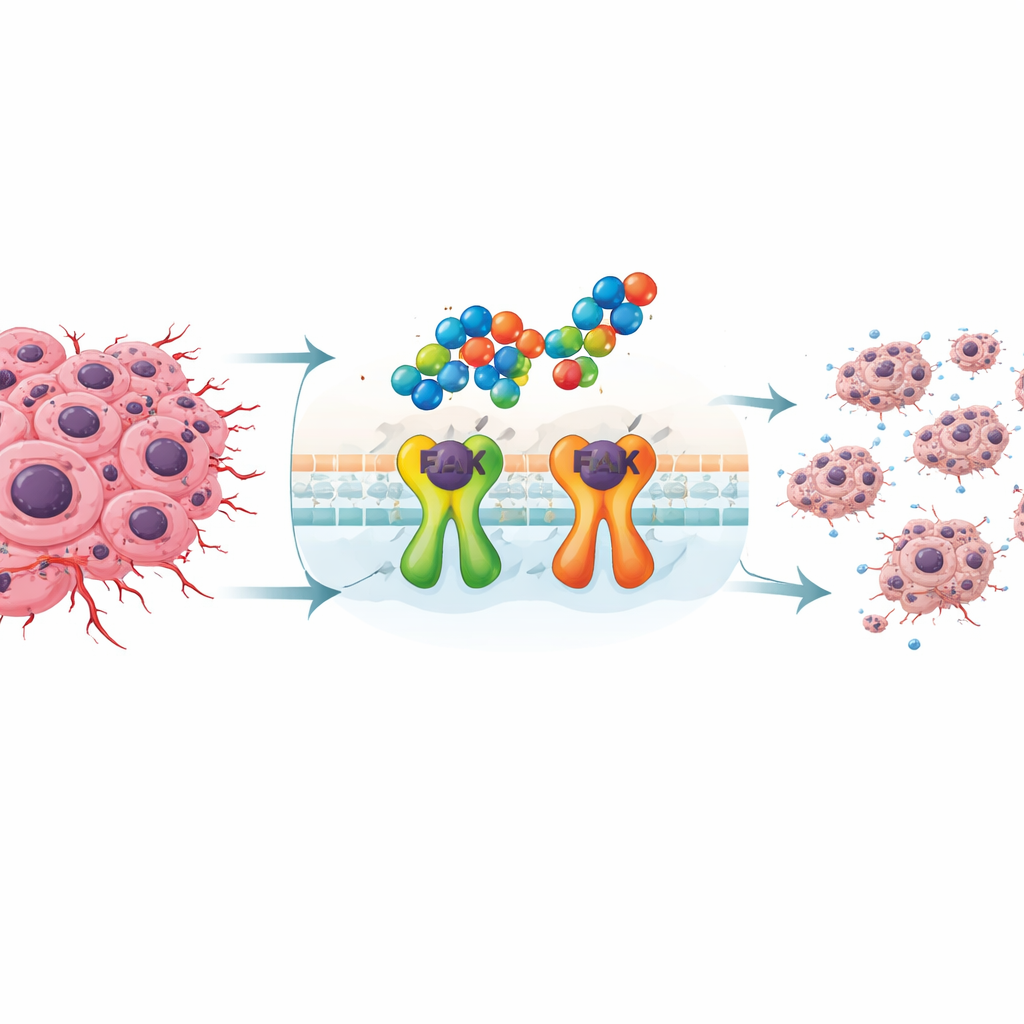
Wachstumshemmung, Bewegungsblockade und Auslösen von Zelltod
Labortests zeigten, dass diese neuen Verbindungen mehr bewirken als nur die Zellteilung zu verlangsamen. In Scratch‑“Wund”‑Experimenten, bei denen in eine Zellschicht eine Lücke gebracht wird und die Forschenden beobachten, wie schnell Zellen diese schließen, verzögerten die Verbindungen 3c, 4c und 5c die Migration in beiden Brustkrebszelllinien deutlich. Verbindung 4c führte Krebszellen außerdem an bestimmten Kontrollpunkten des Zellzyklus zu einem Stillstand: Sie blockierte eine Brustkrebszelllinie (MCF‑7) in der DNA‑Replikationsphase und eine andere, triple‑negative Linie (MDA‑MB‑231), vor der Zellteilung, wodurch ihre Vermehrung eingeschränkt wurde. Beim Blick auf Marker programmierter Zellsterblichkeit zeigte sich, dass 4c bei den triple‑negativen Zellen besonders wirksam war und fast 91 % von ihnen in die Apoptose trieb, mit sehr wenig nekrotischen Schäden — ein Muster, das für therapeutische Zwecke meist wünschenswerter ist.
Den Überlebensschalter des Krebses treffen
Um zu verstehen, wie diese Moleküle wirken, maßen die Forschenden Veränderungen in wichtigen Genen und Proteinen. In den triple‑negativen Zellen senkte 4c die Aktivität des Gens, das FAK kodiert, deutlich und reduzierte die FAK‑Proteinmenge um etwa 61 %, womit es fast mit einem bekannten FAK‑blocker im Vergleich mithalten konnte. Zugleich verringerte 4c Signale, die Zellen vor dem Tod schützen, wie BCL2 und den Chemokin CCL5, und erhöhte pro‑apoptotische Signale wie Caspase‑3. Da FAK und CCL5 auch mit Zellmigration und Immunflucht in Verbindung stehen, hilft ihre Unterdrückung, die starke Anti‑Migrations‑ und Pro‑Apoptose‑Wirkung im Labor zu erklären. Computersimulierte Docking‑Studien stützten dieses Bild, indem sie zeigten, dass 4c gut in die aktive Tasche von FAK passt und mehrere stabilisierende Kontakte eingeht — konsistent mit einer direkten Hemmung des Enzyms.
Erste Sicherheitssignale und nächste Schritte
Bevor ein Wirkstoffkandidat in Richtung klinischer Tests gehen kann, muss seine Sicherheit bewertet werden. Das Team verabreichte Mäusen Einzeldosen von 4c und beobachtete sie zwei Tage lang. Bei der niedrigsten getesteten Dosis von 12,5 mg pro Kilogramm Körpergewicht zeigten die Tiere keine offensichtlichen Anzeichen von Unwohlsein, und Leber‑ sowie Nierengewebe blieben unter dem Mikroskop weitgehend intakt. Höhere Dosen führten zu deutlich sichtbaren Gewebeschäden und biochemischen Stressanzeichen, was auf die Notwendigkeit einer sorgfältigen Dosiswahl hinweist. Zusätzliche Computermodelle zu Resorption, Metabolismus und Toxizität deuteten darauf hin, dass 4c im Vergleich zu einigen bestehenden Krebsmedikamenten insgesamt günstige Eigenschaften besitzt, wobei potenzielle Leber‑ und herzbezogene Risiken jedoch genau überwacht werden müssen. 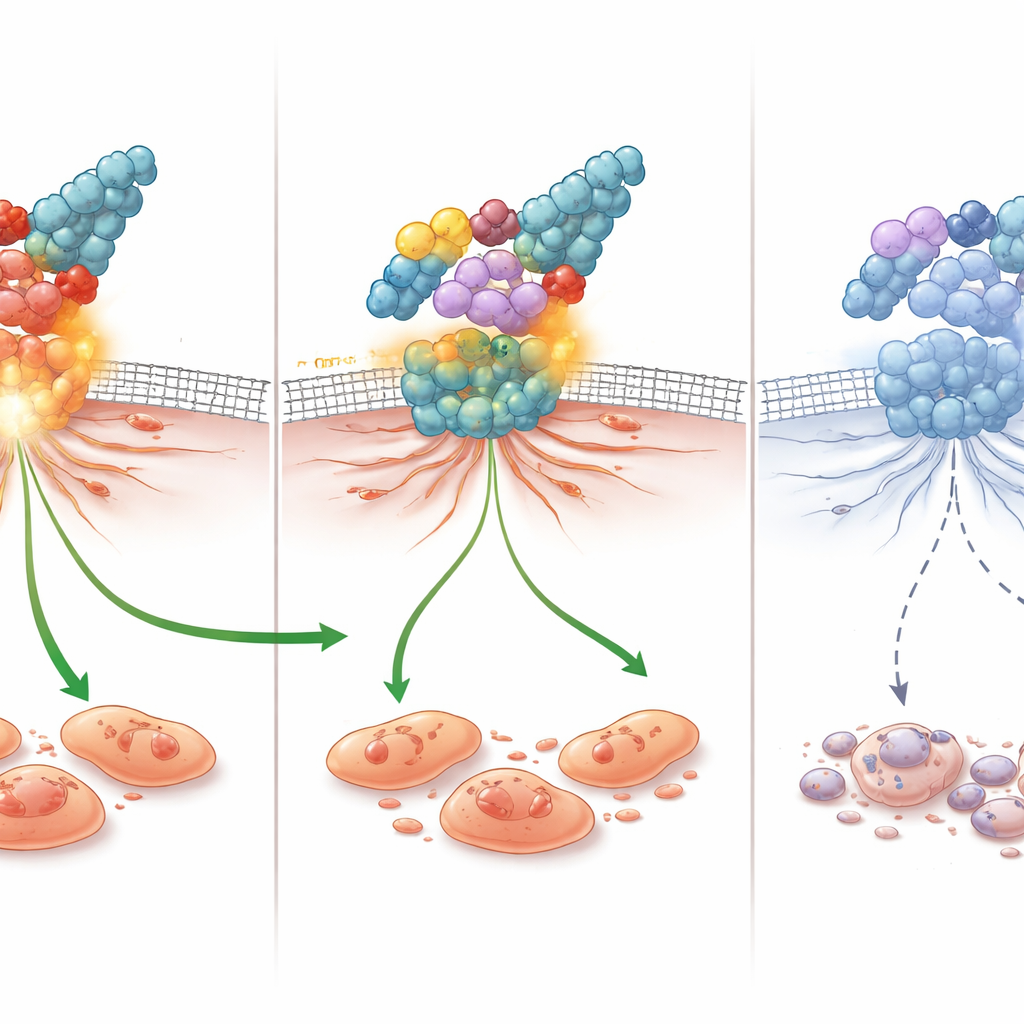
Was das für künftige Behandlungen bedeuten könnte
Kurz gefasst identifiziert diese Arbeit ein neues niedermolekulares Molekül, 4c, das sowohl einen zentralen Überlebensweg in Zellen des triple‑negativen Brustkrebses blockieren als auch diese gezielt in einen geordneten Zelltod treiben kann, während es gleichzeitig deren Bewegungs‑ und Ausbreitungsfähigkeit hemmt. Obwohl die Forschung noch in einem frühen, präklinischen Stadium steht und wesentlich mehr Prüfungen in Tiermodellen mit Tumoren sowie Langzeitsicherheitsstudien erforderlich sind, bietet 4c einen vielversprechenden Entwurf für eine neue Klasse zielgerichteter Therapien gegen eine der herausforderndsten Formen des Brustkrebses.
Zitation: Abd El Salam, H.A., Abu-Shahba, N., Ibrahim Fouad, G. et al. New indole-linked 1,2,4-triazole derivatives as dual FAK inhibitors and apoptosis inducers targeting survival and migration in triple-negative breast cancer in-vitro. Sci Rep 16, 13134 (2026). https://doi.org/10.1038/s41598-026-41032-1
Schlüsselwörter: triple-negativer Brustkrebs, Focal‑Adhesion‑Kinase, zielgerichtete Therapie, Apoptose, niedermolekularer Inhibitor