Clear Sky Science · de
TBL1X/TBL1XR1 steuern die Identität von β‑Zellen über ein PAX6‑enthaltendes Genregulationsnetzwerk
Warum Diabetes ein Problem der Zell‑Identität sein kann
Die meisten Menschen denken bei Diabetes an einen Fehler im Blutzuckerstoffwechsel, doch im Inneren der Bauchspeicheldrüse geht es eigentlich um Identität. Spezialisierte Beta‑Zellen erkennen steigenden Zucker und schütten Insulin aus. Bei Typ‑2‑Diabetes verlieren viele dieser Zellen ihren ausgereiften Charakter und erfüllen ihre Aufgabe nicht mehr richtig. Diese Studie deckt eine bislang fehlende Kontrollschicht auf, die Beta‑Zellen hilft, ihrer Identität treu zu bleiben, und zeigt, wie ihr Versagen den Körper in Richtung Diabetes kippen kann.
Die Wächter innerhalb der Beta‑Zellen
Beta‑Zellen leben in kleinen Gruppen in der Bauchspeicheldrüse, den Langerhans‑Inseln, und sind für die Produktion und Sekretion von Insulin verantwortlich. Ihre Identität wird von „Master‑Schaltern“ in der DNA bestimmt, den Transkriptionsfaktoren, die die richtigen Gene an- und die falschen ausschalten. Diese Master‑Schalter wirken jedoch nicht allein; sie sind auf Helferproteine angewiesen, die feinabstimmen, wann Gene aktiviert oder stillgelegt werden. Die Autoren konzentrieren sich auf zwei solche Helfer, TBL1X und TBL1XR1 (zusammen TBL/R1 genannt), mit der Frage, ob sie für die Aufrechterhaltung der Beta‑Zellfunktion unerlässlich sind und welche Bedeutung sie für den Menschen mit Diabetes haben.
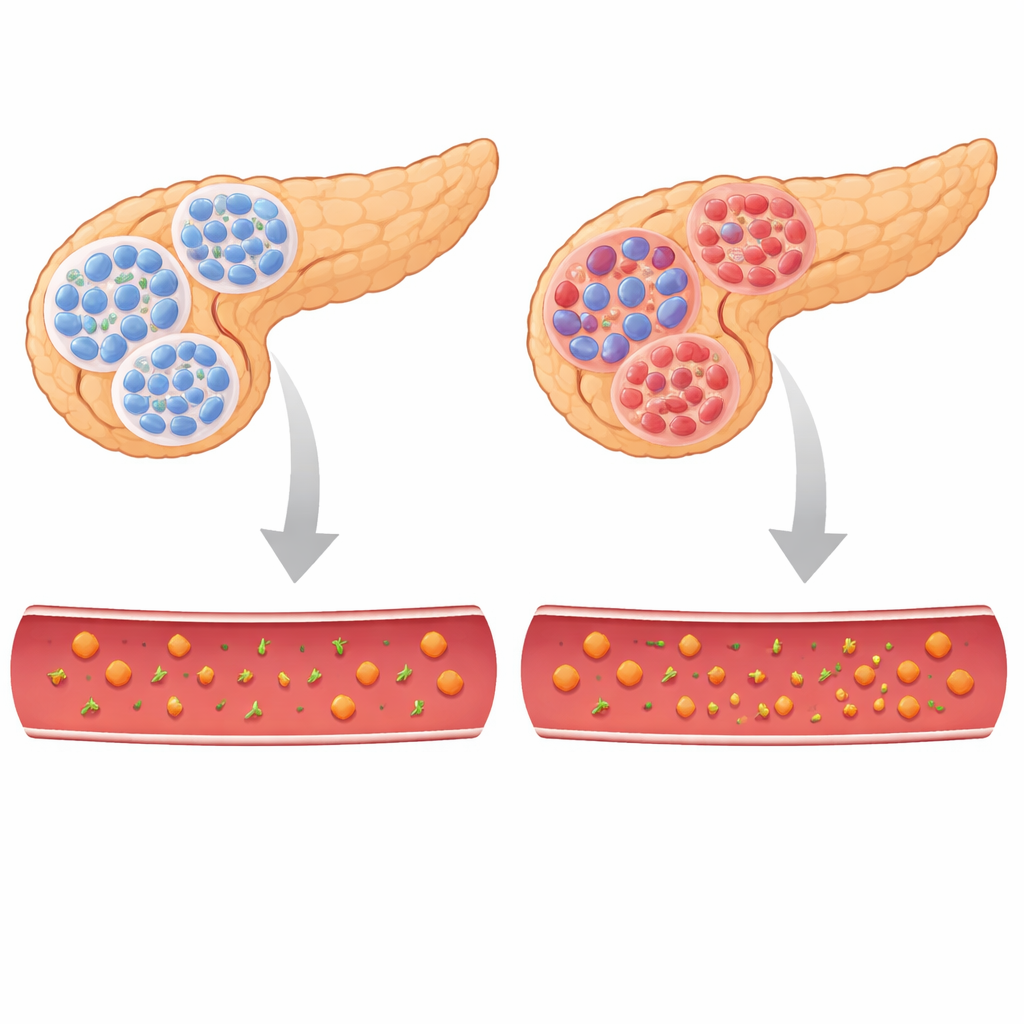
Was passiert, wenn die Wächter entfernt werden
Um die Rolle von TBL/R1 zu prüfen, erzeugten die Forscher Mäuse, in denen diese Helfer spezifisch in Beta‑Zellen gelöscht wurden. Zunächst wirkten die jungen Tiere normal, doch bald entwickelten sie sehr hohe Blutzuckerwerte und Gewichtsverlust – ein klassisches diabetes‑ähnliches Bild. Ihr Körper war nicht insulinresistent; vielmehr produzierte die Bauchspeicheldrüse schlicht zu wenig Insulin. Messungen zeigten niedrige Insulinspiegel im Blut und reduzierte Insulinreserven in der Bauchspeicheldrüse, während die Spiegel des Hormons Glukagon, das den Blutzucker erhöht, anstiegen. Schon bevor der Blutzucker stieg, war die Struktur der Inseln gestört: Es gab weniger Beta‑Zellen, Alpha‑Zellen waren über die Inseln verstreut, und das Verhältnis dieser Zelltypen ähnelte dem, das bei diabetischen Tieren beobachtet wird.
Zellen, die vergessen, wer sie sind
Bei genauerer Betrachtung der Genaktivität in den Inseln fanden die Forscher weitreichende Veränderungen. Tausende Gene änderten ihr Verhalten, wenn TBL/R1 fehlten. Wichtige Marker ausgereifter Beta‑Zellen, darunter die Gene für Insulin und Proteine, die für die Erkennung und Verarbeitung von Glukose erforderlich sind, wurden herunterreguliert. Gleichzeitig stiegen Gene an, die in gesunden Beta‑Zellen normalerweise „nicht erlaubt“ sind, ebenso wie Marker unreifer oder alternativer Inselzelltypen. Einzelzell‑RNA‑Sequenzierung, die einzelne Zellen profiliert, bestätigte, dass echte Beta‑Zellen verloren gingen und durch weniger ausgereifte Zellen sowie seltene „polyhormonale“ Zellen ersetzt wurden, die eine Mischung von Hormonen produzierten. Diese Muster deuten darauf hin, dass Beta‑Zellen ohne TBL/R1 nicht unbedingt absterben; vielmehr entfernen sie sich von ihrer spezialisierten Identität hin zu einem primitiveren oder gemischten Zustand, der keine normale Insulinproduktion mehr aufrechterhält.
Ein molekulares Zusammenspiel zwischen PAX6 und TBL/R1
Um den zugrunde liegenden Mechanismus zu verstehen, kartierten die Forscher, welche Kernproteine physisch mit TBL/R1 in Beta‑Zelllinien interagieren. Unter vielen Partnern identifizierten sie PAX6, einen bekannten Regulator der Inselentwicklung und funktion. Sie zeigten, dass PAX6 und TBL/R1 gemeinsam mit einem weiteren Protein, HDAC3, im Kontrollbereich des Insulin‑Gens sitzen und dort ein regulatorisches Team direkt auf der DNA bilden. Wenn TBL/R1 experimentell reduziert wurden, konnte PAX6 die Aktivität des Insulin‑Gens nicht mehr steigern, und Insulinproduktion sowie Sekretion sanken. Die Hemmung von HDAC3 schwächte ebenfalls die Aktivierung des Insulin‑Gens in einer von TBL/R1 abhängigen Weise, was darauf hindeutet, dass dieses Trio als Schalter wirkt, der die Insulin‑Genexpression fördert und gleichzeitig unangemessene Gene unterdrückt.
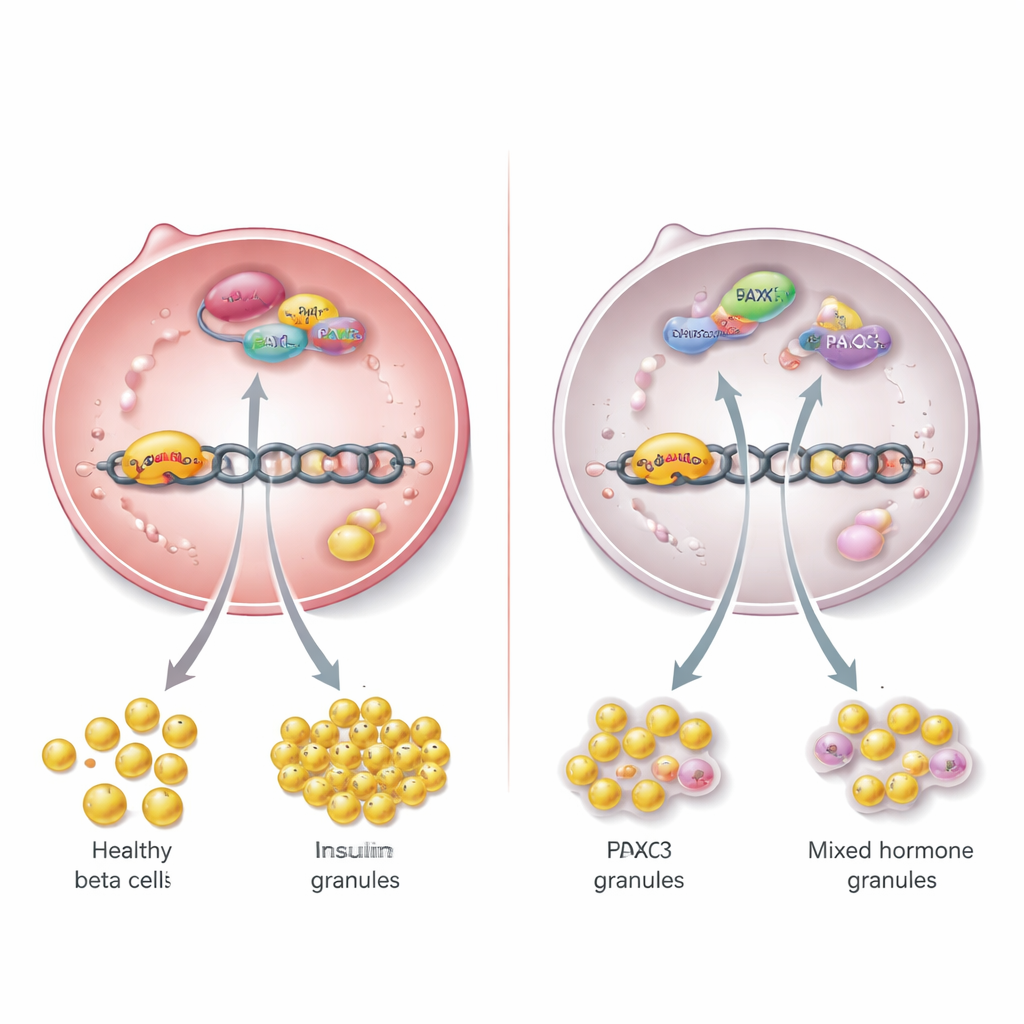
Vom Mausmechanismus zum menschlichen Risiko
Wichtig ist, dass dieses Regulationssystem nicht auf Nagetiere beschränkt ist. In einer menschlichen Beta‑Zelllinie fanden sich TBL1X, TBL1XR1 und PAX6 ebenfalls am menschlichen Insulin‑Gen und bildeten ähnliche Komplexe mit HDAC3. Die Reduktion von TBL/R1 in diesen Zellen verringerte die Insulin‑Genaktivität und die Insulinsekretion. Die Forscher untersuchten dann menschliche Spender‑Inseln und fanden, dass eine niedrigere TBL1X‑Expression mit höheren Langzeit‑Blutzuckerwerten, gemessen am HbA1c, assoziiert war. Große genetische Studien an Zehntausenden Menschen zeigten darüber hinaus, dass häufige DNA‑Varianten in der Nähe der TBL1X‑ und TBL1XR1‑Gene mit erhöhtem HbA1c und erhöhtem zufälligem Blutzucker verbunden sind, wodurch diese Regulationsschicht mit dem Diabetesrisiko in der Bevölkerung verknüpft wird.
Warum diese Ergebnisse für zukünftige Therapien wichtig sind
Insgesamt zeigt die Studie, dass TBL1X und TBL1XR1 als entscheidende Wächter der Beta‑Zell‑Identität wirken, indem sie mit PAX6 und assoziierten Komplexen zusammenarbeiten, um Insulin‑Gene aktiv zu halten und unangemessene Gene stummzulegen. Wenn diese Kontrollschicht versagt, vergessen Beta‑Zellen nach und nach, wer sie sind, was zu zu wenig Insulin und steigendem Blutzucker führt. Da der Verlust der Beta‑Zell‑Identität reversibel sein kann, könnte das Ansteuern dieses auf TBL/R1 zentrierten Netzwerks – entweder zum Schutz bestehender Beta‑Zellen oder zur Verbesserung der Reifung im Labor gezüchteter Zellen für die Transplantation – neue, gezieltere Behandlungsstrategien für Typ‑2‑Diabetes eröffnen.
Zitation: Walth-Hummel, A.A., Jouffe, C., Weber, P. et al. TBL1X/TBL1XR1 govern β-cell identity through a PAX6-containing gene regulatory network. Nat Commun 17, 3736 (2026). https://doi.org/10.1038/s41467-026-72077-5
Schlüsselwörter: Beta‑Zell‑Identität, Insulin‑Regulation, transkriptionelle Kofaktoren, Typ‑2‑Diabetes, PAX6‑Netzwerk