Clear Sky Science · sv
Utvärdering av ensemblelärande för detektion av horisontell genöverföring
Varför detta spelar roll för bakterier och läkemedel
Bakterier kan byta användbara gener som samlarkort, vilket gör att de snabbt kan få egenskaper som antibiotikaresistens. Många av dessa införskaffade gener ligger i särskilda kluster i genomet som kallas genomöar. Att upptäcka dessa öar mer tillförlitligt kan stärka insatser för att spåra och kontrollera antimikrobiell resistens. Denna studie undersöker om sammanslagning av flera maskininlärningsvyer av DNA till ett enda ”ensemble” kan förbättra upptäckten av dessa öar och vad det innebär för hur vi utformar sådana verktyg.
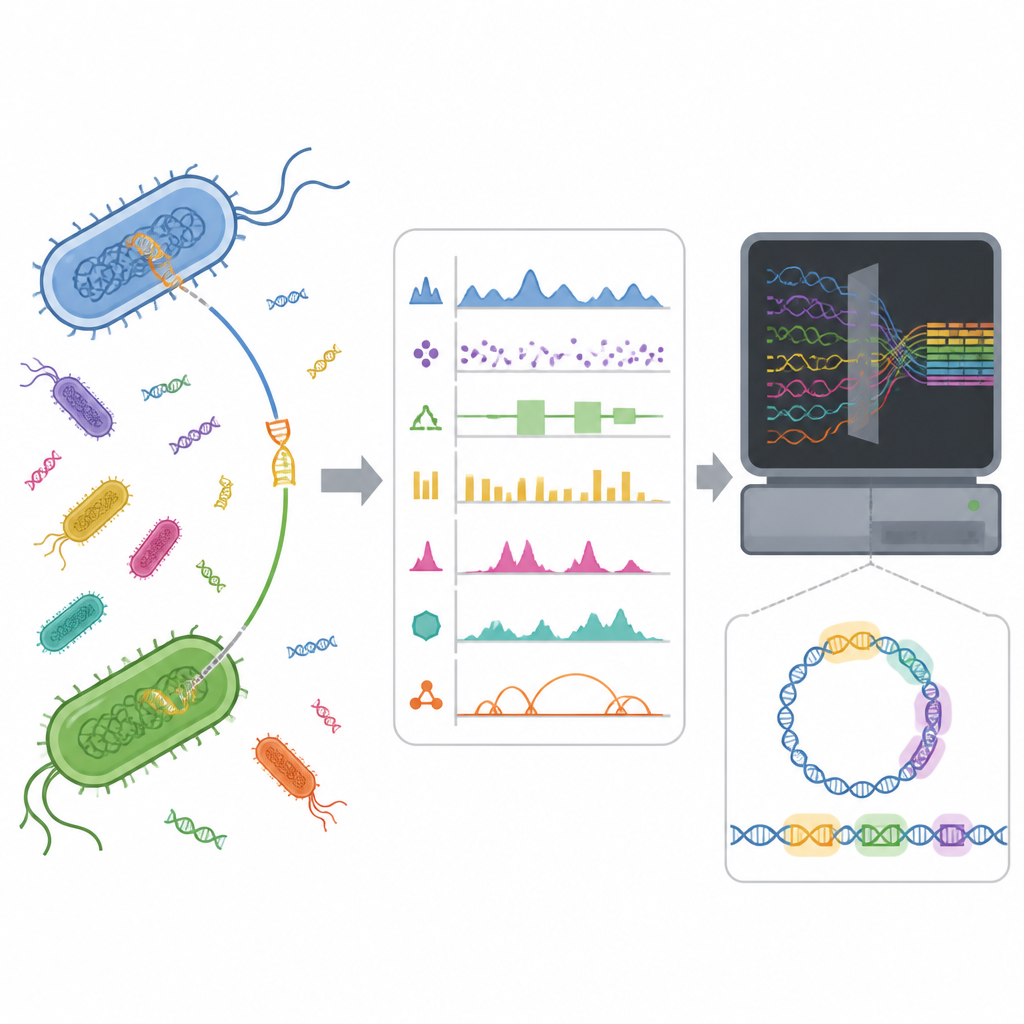
Dolda DNA-öar i bakteriella genom
Bakterier förlitar sig inte endast på långsam mutation över generationer. De tar ofta emot färdiga genetiska paket från andra mikrober genom horisontell genöverföring. Dessa paket, kända som genomöar, kan bära gener för virulens, överlevnad i hårda miljöer eller resistens mot antibiotika. Att hitta dessa öar i ett genom är utmanande eftersom de kan anta många former och smälta in i värdens DNA. Bättre detektion kan hjälpa forskare att förstå hur skadliga egenskaper sprids och stödja folkhälsans övervakning av antimikrobiell resistens.
Att lära datorer att upptäcka ovanligt DNA
Beräkningsverktyg försöker markera genomöar genom att leta efter ovanliga mönster i DNA-sekvensen eller genom att jämföra genom. Nyare maskininlärningsmetoder representerar samma DNA-segment på många olika sätt, till exempel genom att räkna korta sekvensfragment eller sammanfatta kemiska egenskaper. Tidigare arbete av författarna visade att medan en representation fungerade bäst överlag, fångade flera andra med låg korrelation olika men lika användbara signaler. Detta antydde att en kombination av dessa olika vyer skulle kunna hjälpa en modell att känna igen genomöar mer fullständigt än någon enskild vy ensam.
Bygga modellteam istället för en expert
Forskarna testade idén genom att skapa ett ensemble av modeller tränade på 44 olika DNA-representationer med fem vanliga klassificerare. De valde först den bästa modellen för varje representation, och använde sedan en tvåstegsprocess för att välja kombinationer som var både precisa och olika i sina förutsägelser. Flera ensemble-strategier prövades, inklusive enkel omröstning och en mer lagerad stacking-ansats där en separat modell lär sig hur de andra ska kombineras. På en referenssamling av bakteriella DNA-segment förbättrade de bästa ensamblen mått som återkallning något, vilket betyder att de fångade fler genomöar än den bästa enskilda modellen, även om vinsterna var måttliga och inte statistiskt starka.
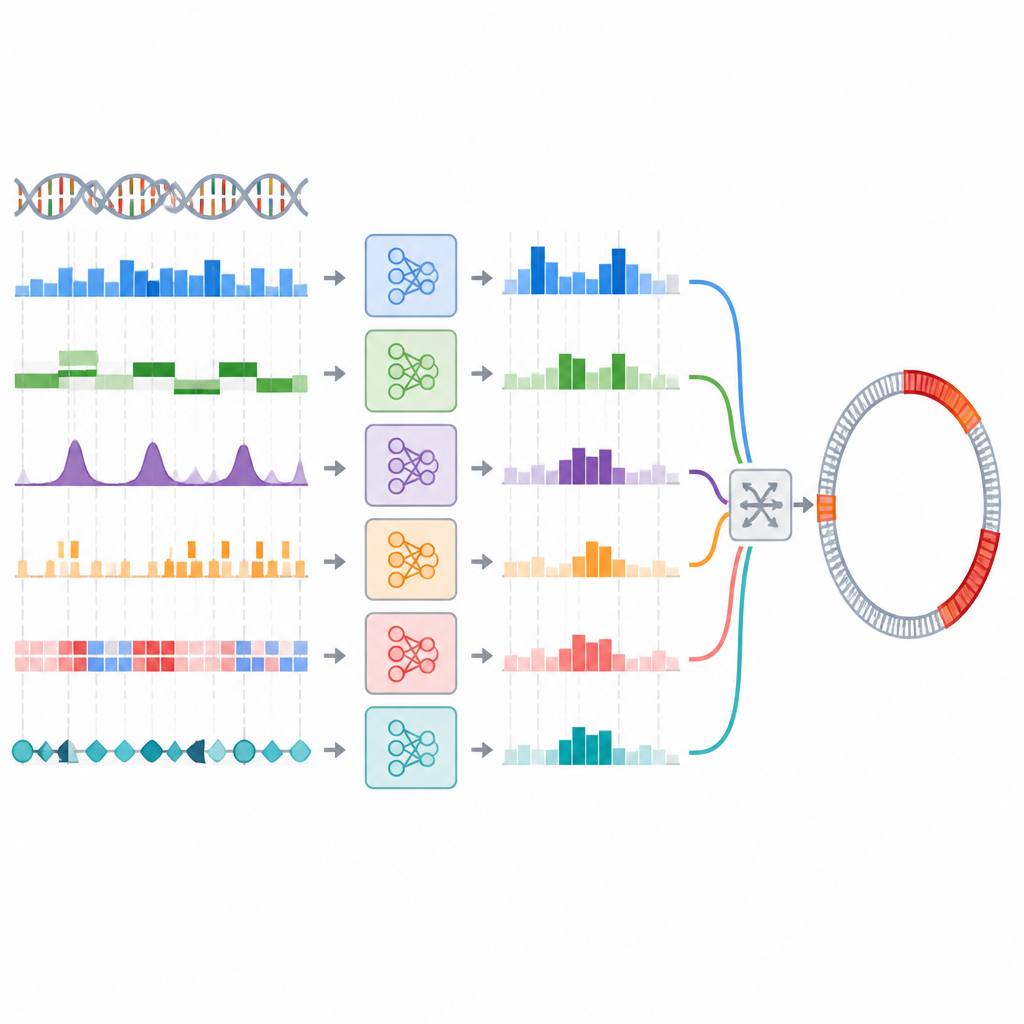
Från segmentsetiketter till verkliga genomkartor
I praktiken behöver forskare inte bara märka korta DNA-fragment utan också kartlägga de exakta gränserna för genomöar längs hela genom. Gruppen testade om deras ensemble som presterade bra i segmentklassificeringsuppgiften också skulle förbättra dessa gränsprognoser när den kopplades in i en befintlig genomskanningspipeline. Här förändrades bilden. Ett omröstningsbaserat ensemble hade svårt och missade många öar om inte trösklar justerades noggrant, och även då nådde det inte upp till den enskilt bästa modellen. Ett stacking-baserat ensemble presterade ungefär lika väl som den enskilda modellen men överträffade den inte tydligt. Sammantaget översatte de sofistikerade ensamblen inte sin lilla klassificeringsfördel till bättre kartläggning över hela genomet.
Ompröva hur vi formulerar problemet
Författarna drar slutsatsen att kombination av olika DNA-representationer kan hjälpa modeller att uppmärksamma fler kandidatgenomöar, men förbättringen är begränsad och känslig för hur förutsägelser används. Viktigare är att studien visar att träning av modeller enbart för att klassificera förutbestämda DNA-segment inte är tillräckligt när det verkliga målet är att rita exakta ögränser över kompletta genom. Arbetet förespråkar att omdefiniera detektion av genomöar som ett verkligt genomskannings- eller till och med regressionsproblem, underbyggt av bättre benchmark-dataset och kontextkänsliga modeller. Fram till dess förblir nuvarande pipelines användbara men måste tillämpas med försiktighet när de används för att informera studier om spridning av antibiotikaresistens.
Citering: Wijaya, A.J., Anžel, A. & Hattab, G. Evaluating ensemble learning approaches for horizontal gene transfer detection. Sci Rep 16, 16582 (2026). https://doi.org/10.1038/s41598-026-53037-x
Nyckelord: horisontell genöverföring, genomöar, ensemblelärande, antimikrobiell resistens, maskininlärning genomik