Clear Sky Science · it
Valutazione degli approcci di ensemble learning per il rilevamento del trasferimento genico orizzontale
Perché questo è importante per germi e farmaci
I batteri possono scambiarsi geni utili come fossero carte da collezione, permettendo loro di acquisire rapidamente tratti come la resistenza agli antibiotici. Molti di questi geni acquisiti risiedono in cluster particolari nel genoma chiamati isole genomiche. Individuare queste isole in modo più affidabile potrebbe rafforzare gli sforzi per tracciare e controllare la resistenza antimicrobica. Questo studio esplora se combinare diverse viste del DNA tramite machine learning in un unico “ensemble” possa migliorare il rilevamento di tali isole e cosa ciò implichi per la progettazione di questi strumenti.
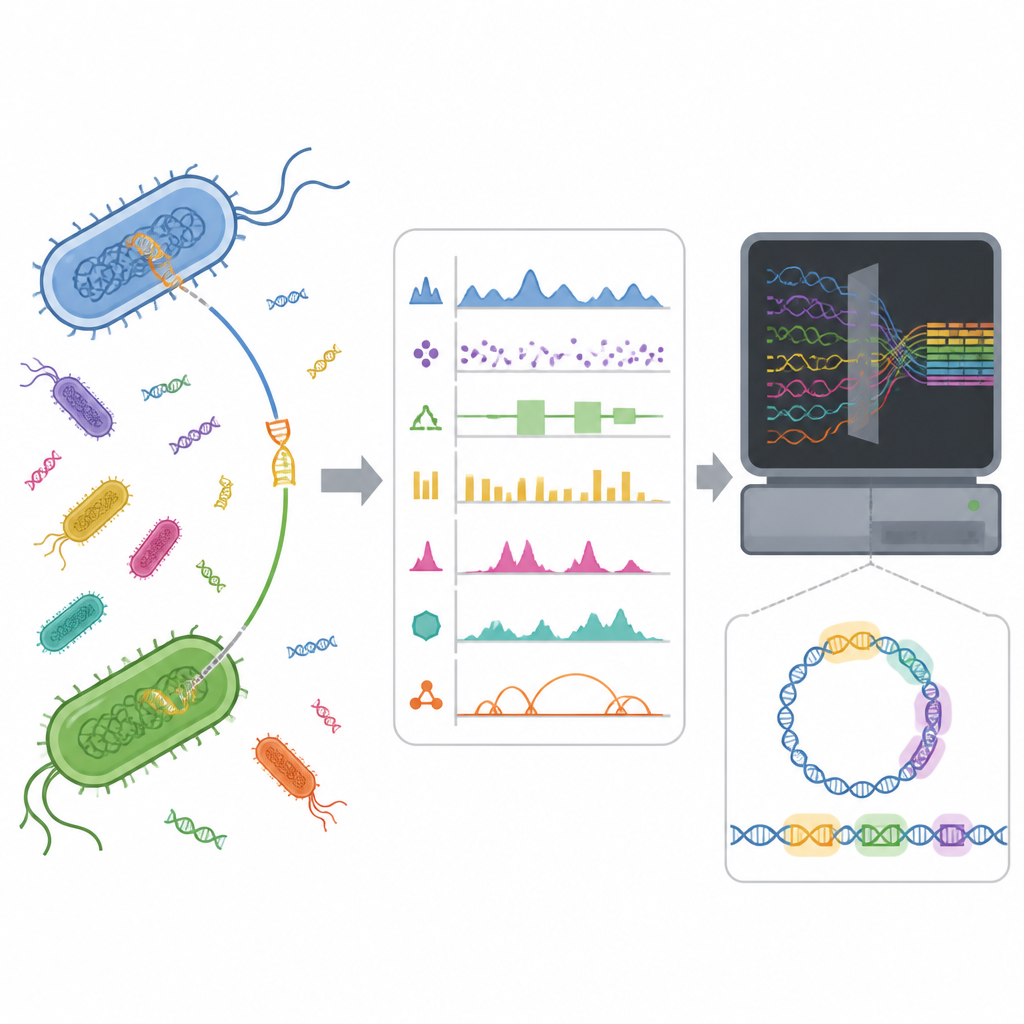
Isole di DNA nascoste nei genomi batterici
I batteri non si affidano solo alla lenta mutazione attraverso le generazioni. Spesso acquisiscono pacchetti genetici già pronti da altri microrganismi tramite trasferimento genico orizzontale. Questi pacchetti, noti come isole genomiche, possono portare geni per la virulenza, la sopravvivenza in ambienti avversi o la resistenza agli antibiotici. Individuare queste isole in un genoma è impegnativo perché si presentano in molte forme e possono confondersi con il DNA dell’ospite. Un rilevamento migliore può aiutare i ricercatori a capire come si diffondono i tratti dannosi e supportare la sorveglianza pubblica della resistenza antimicrobica.
Insegnare ai computer a riconoscere il DNA anomalo
Gli strumenti computazionali cercano di segnalare le isole genomiche osservando pattern insoliti nella sequenza del DNA o confrontando genomi. Metodi recenti di machine learning rappresentano lo stesso segmento di DNA in molti modi diversi, ad esempio contando brevi frammenti di sequenza o riassumendo proprietà chimiche. Lavori precedenti degli autori hanno mostrato che, mentre una rappresentazione si comportava meglio in termini globali, altre varie e debolmente correlate catturavano segnali diversi ma ugualmente utili. Questo suggeriva che combinare queste diverse viste avrebbe potuto aiutare un modello a riconoscere le isole genomiche in modo più completo rispetto a una singola vista.
Costruire squadre di modelli invece di un solo esperto
I ricercatori hanno testato questa idea creando un ensemble di modelli addestrati su 44 diverse rappresentazioni del DNA utilizzando cinque classificatori comuni. Hanno prima scelto il modello migliore per ciascuna rappresentazione, poi hanno usato un processo in due fasi per selezionare combinazioni accurate e diverse nelle loro predizioni. Sono state provate diverse strategie di ensemble, tra cui un semplice voto e un approccio a più livelli (stacking) in cui un modello separato impara a combinare gli altri. Su una raccolta di riferimento di segmenti di DNA batterico, i migliori ensemble hanno migliorato lievemente metriche come il richiamo (recall), ovvero hanno rilevato più isole genomiche rispetto al miglior modello singolo, sebbene i guadagni fossero modesti e non statisticamente solidi.
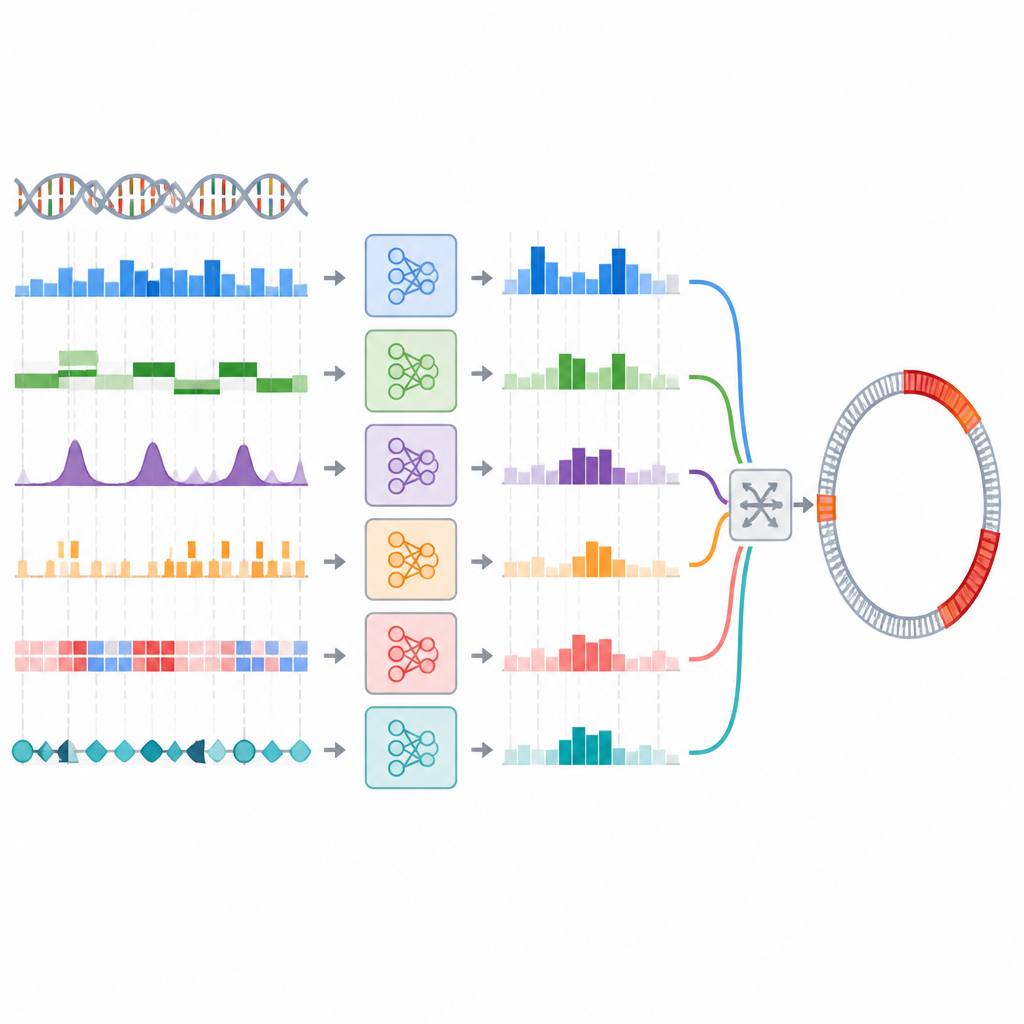
Dalle etichette dei segmenti alle mappe reali del genoma
Nell’uso pratico, gli scienziati hanno bisogno non solo di etichettare brevi frammenti di DNA ma anche di mappare i confini esatti delle isole genomiche lungo interi genomi. Il team ha verificato se l’ensemble che aveva performato bene nel compito di classificazione dei segmenti avrebbe migliorato anche queste predizioni dei confini una volta integrato in una pipeline di scansione del genoma esistente. Qui la situazione è cambiata. Un ensemble basato sul voto ha faticato, perdendo molte isole a meno che le soglie non fossero regolate con cura, e anche in quel caso non ha raggiunto il modello singolo migliore. Un ensemble basato su stacking ha avuto prestazioni paragonabili al modello singolo ma non lo ha superato chiaramente. Nel complesso, gli ensemble sofisticati non hanno tradotto il loro piccolo vantaggio nella classificazione in un miglioramento della mappatura a livello genomico.
Ripensare il modo di inquadrare il problema
Gli autori concludono che combinare diverse rappresentazioni del DNA può aiutare i modelli a individuare più potenziali isole genomiche, ma il miglioramento è limitato e dipende da come vengono usate le predizioni. Più importante, lo studio mostra che addestrare i modelli solo per classificare segmenti di DNA pre-tagliati non è sufficiente quando l’obiettivo reale è tracciare confini accurati delle isole su genomi completi. Il lavoro sostiene la necessità di ridefinire il rilevamento delle isole genomiche come un vero problema di scansione del genoma o addirittura di regressione, supportato da dataset di riferimento migliori e modelli sensibili al contesto. Fino ad allora, le pipeline attuali restano utili ma devono essere applicate con cautela quando informano studi sulla diffusione della resistenza agli antibiotici.
Citazione: Wijaya, A.J., Anžel, A. & Hattab, G. Evaluating ensemble learning approaches for horizontal gene transfer detection. Sci Rep 16, 16582 (2026). https://doi.org/10.1038/s41598-026-53037-x
Parole chiave: trasferimento genico orizzontale, isole genomiche, ensemble learning, resistenza antimicrobica, machine learning genomica