Clear Sky Science · pl
Ocena podejść uczenia zespołowego do wykrywania poziomego transferu genów
Dlaczego to ma znaczenie dla drobnoustrojów i leków
Bakterie mogą wymieniać użyteczne geny niczym karty kolekcjonerskie, co pozwala im szybko zdobywać cechy, takie jak oporność na antybiotyki. Wiele z tych zapożyczonych genów znajduje się w specjalnych skupiskach w genomie zwanych wyspami genomowymi. Bardziej niezawodne wykrywanie tych wysp mogłoby wzmocnić wysiłki mające na celu monitorowanie i kontrolę oporności na środki przeciwdrobnoustrojowe. W tym badaniu zbadano, czy połączenie kilku reprezentacji DNA w jednym „zespole” modeli może poprawić wykrywanie tych wysp oraz jakie są tego implikacje dla projektowania takich narzędzi.
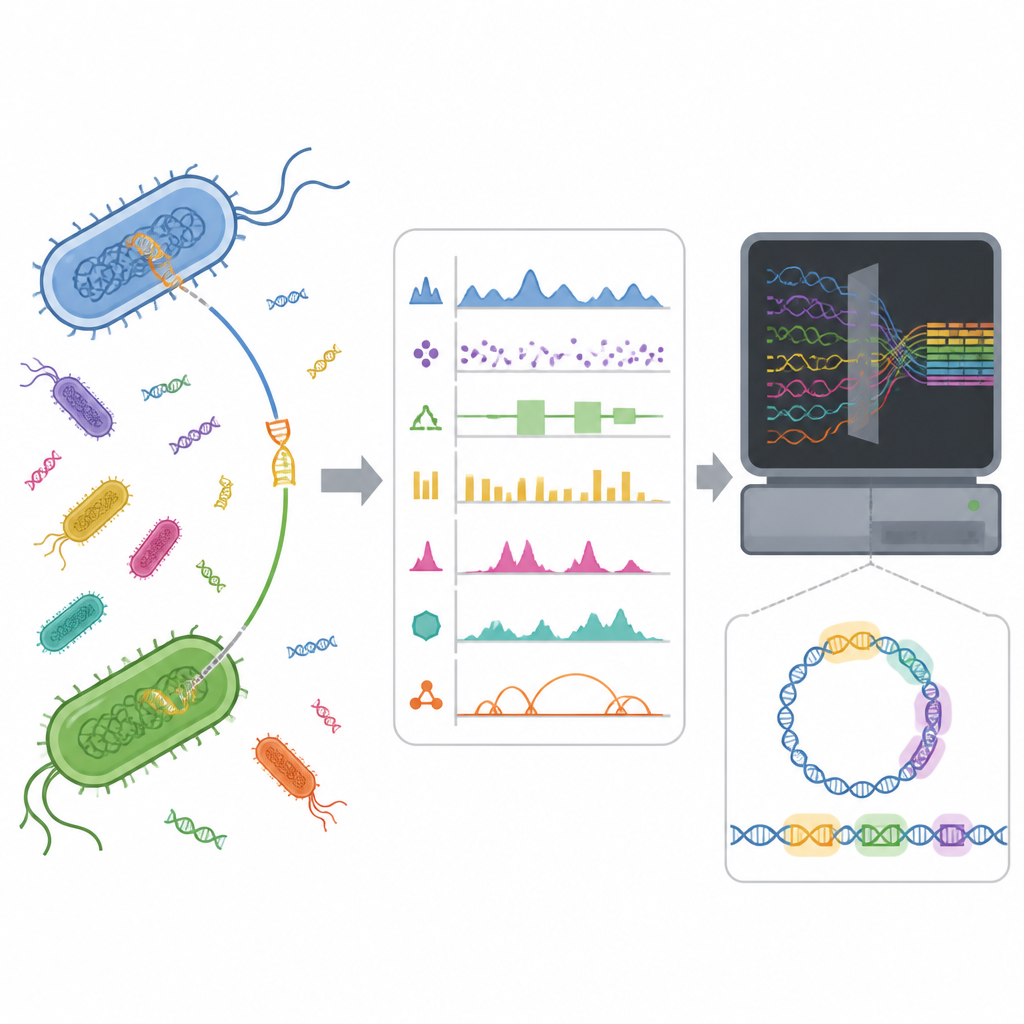
Ukryte wyspy DNA w genomach bakterii
Bakterie nie polegają wyłącznie na powolnych mutacjach w kolejnych pokoleniach. Często pozyskują gotowe pakiety genetyczne od innych mikrobów poprzez poziomy transfer genów. Te pakiety, znane jako wyspy genomowe, mogą zawierać geny odpowiadające za zjadliwość, przetrwanie w trudnych warunkach lub oporność na antybiotyki. Znalezienie tych wysp w genomie jest trudne, ponieważ występują w różnych formach i mogą wtopić się w DNA gospodarza. Lepsze wykrywanie może pomóc badaczom zrozumieć, jak rozprzestrzeniają się szkodliwe cechy, oraz wesprzeć nadzór nad opornością na środki przeciwdrobnoustrojowe.
Nauczanie komputerów rozpoznawania nietypowego DNA
Narzędzia obliczeniowe próbują wskazywać wyspy genomowe, szukając nietypowych wzorców w sekwencji DNA lub porównując genomy. Nowsze metody uczenia maszynowego representują ten sam fragment DNA na wiele sposobów, na przykład licząc krótkie fragmenty sekwencji lub podsumowując właściwości chemiczne. Wcześniejsze prace autorów wykazały, że choć jedna reprezentacja wypadała najlepiej ogólnie, kilka innych o niskiej korelacji uchwyciło różne, ale równie użyteczne sygnały. Sugerowało to, że połączenie tych różnych widoków mogłoby pomóc modelowi rozpoznać wyspy genomowe bardziej kompletnie niż jakikolwiek pojedynczy widok.
Budowanie zespołów modeli zamiast jednego eksperta
Naukowcy przetestowali ten pomysł, tworząc zespół modeli trenowanych na 44 różnych reprezentacjach DNA przy użyciu pięciu powszechnych klasyfikatorów. Najpierw wybrali najlepszy model dla każdej reprezentacji, a następnie zastosowali dwuetapowy proces selekcji kombinacji, które były jednocześnie dokładne i zróżnicowane w swoich przewidywaniach. Przetestowano kilka strategii zespołowych, w tym proste głosowanie i bardziej warstwowe podejście stacking, w którym osobny model uczy się, jak łączyć pozostałe. Na zestawie odniesienia fragmentów DNA bakterii najlepsze zespoły nieznacznie poprawiły miary takie jak recall, co oznacza, że wychwyciły więcej wysp genomowych niż najlepszy pojedynczy model, chociaż zyski były skromne i nie były statystycznie silne.
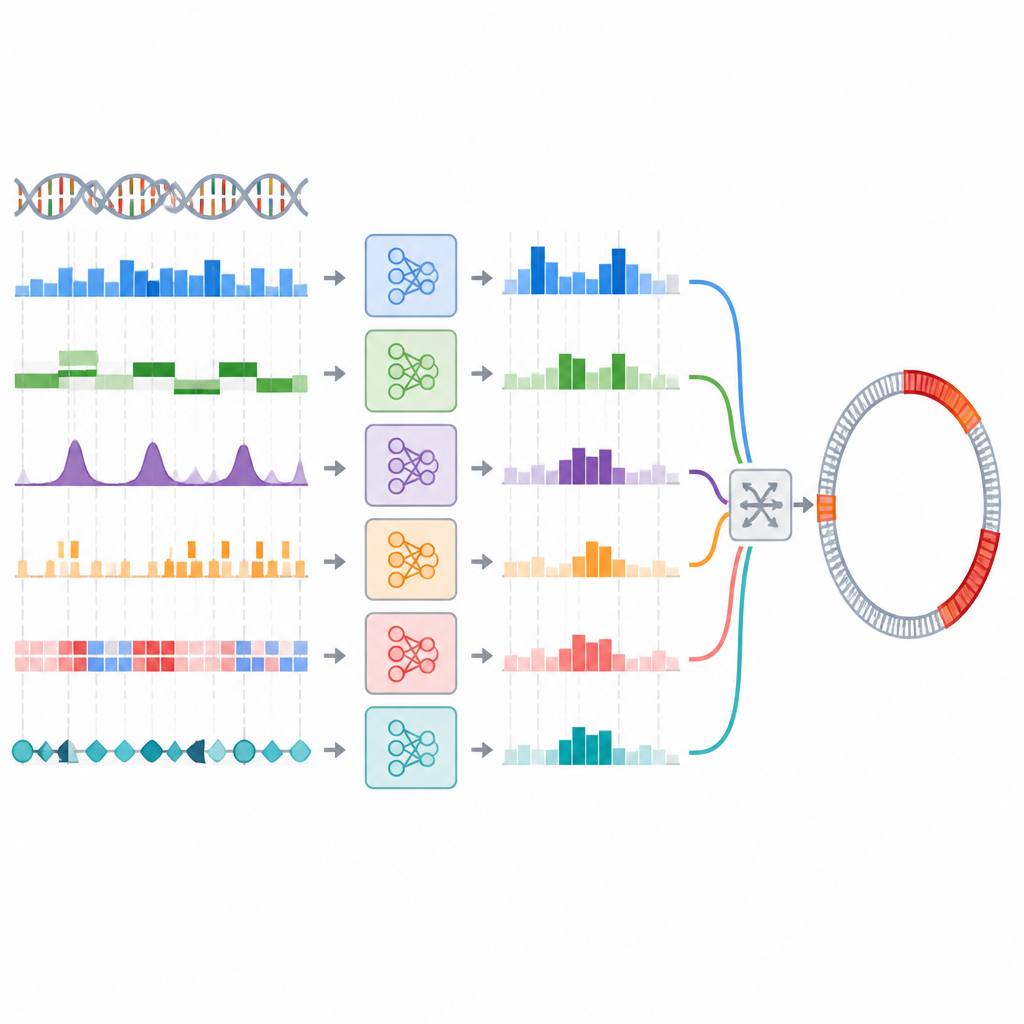
Od etykiet fragmentów do rzeczywistych map genomów
W praktycznym użyciu naukowcy potrzebują nie tylko oznaczania krótkich fragmentów DNA, ale też wyznaczania dokładnych granic wysp genomowych w całych genomach. Zespół sprawdził, czy ich ensemble, który dobrze wypadł w zadaniu klasyfikacji fragmentów, poprawi także przewidywanie granic, gdy zostanie włączony do istniejącego pipeline’u skanującego genomy. Tutaj obraz się zmienił. Ensemble oparty na głosowaniu miał trudności, przegapiając wiele wysp, jeśli progi nie były starannie dostrojone, i nawet wtedy nie dorównywał najlepszemu pojedynczemu modelowi. Ensemble stackingowy wypadł mniej więcej tak samo jak pojedynczy model, ale wyraźnie go nie przewyższył. Ogólnie rzecz biorąc, zaawansowane zespoły nie przełożyły swojej niewielkiej przewagi w klasyfikacji na lepsze mapowanie w skali genomu.
Ponowne przemyślenie sposobu formułowania problemu
Autorzy konkludują, że łączenie różnych reprezentacji DNA może pomóc modelom zauważyć więcej kandydatów na wyspy genomowe, ale poprawa jest ograniczona i wrażliwa na sposób wykorzystania przewidywań. Co ważniejsze, badanie pokazuje, że uczenie modeli wyłącznie do klasyfikacji uprzednio pociętych fragmentów DNA nie wystarcza, gdy prawdziwym celem jest narysowanie dokładnych granic wysp w całych genomach. Praca ta argumentuje za przedefiniowaniem wykrywania wysp genomowych jako prawdziwego problemu skanowania genomu, a nawet regresji, wspieranego lepszymi danymi benchmarkowymi i modelami uwzględniającymi kontekst. Do tego czasu obecne pipeline’y pozostają użyteczne, ale należy je stosować ostrożnie przy informowaniu badań nad rozprzestrzenianiem się oporności na antybiotyki.
Cytowanie: Wijaya, A.J., Anžel, A. & Hattab, G. Evaluating ensemble learning approaches for horizontal gene transfer detection. Sci Rep 16, 16582 (2026). https://doi.org/10.1038/s41598-026-53037-x
Słowa kluczowe: poziomy transfer genów, wyspy genomowe, uczenie zespołowe, oporność na środki przeciwdrobnoustrojowe, genomika uczenia maszynowego