Clear Sky Science · fr
Évaluation des approches d’apprentissage ensembliste pour la détection de transfert horizontal de gènes
Pourquoi cela compte pour les microbes et les médicaments
Les bactéries peuvent échanger des gènes utiles comme des cartes à collectionner, ce qui leur permet d’acquérir rapidement des caractères tels que la résistance aux antibiotiques. Beaucoup de ces gènes empruntés se trouvent dans des regroupements spéciaux du génome appelés îlots génomiques. Repérer ces îlots de manière plus fiable pourrait renforcer les efforts de surveillance et de contrôle de la résistance aux antimicrobiens. Cette étude examine si la combinaison de plusieurs représentations de l’ADN dans un « ensemble » d’algorithmes peut améliorer la détection de ces îlots et quelles en sont les implications pour la conception de tels outils.
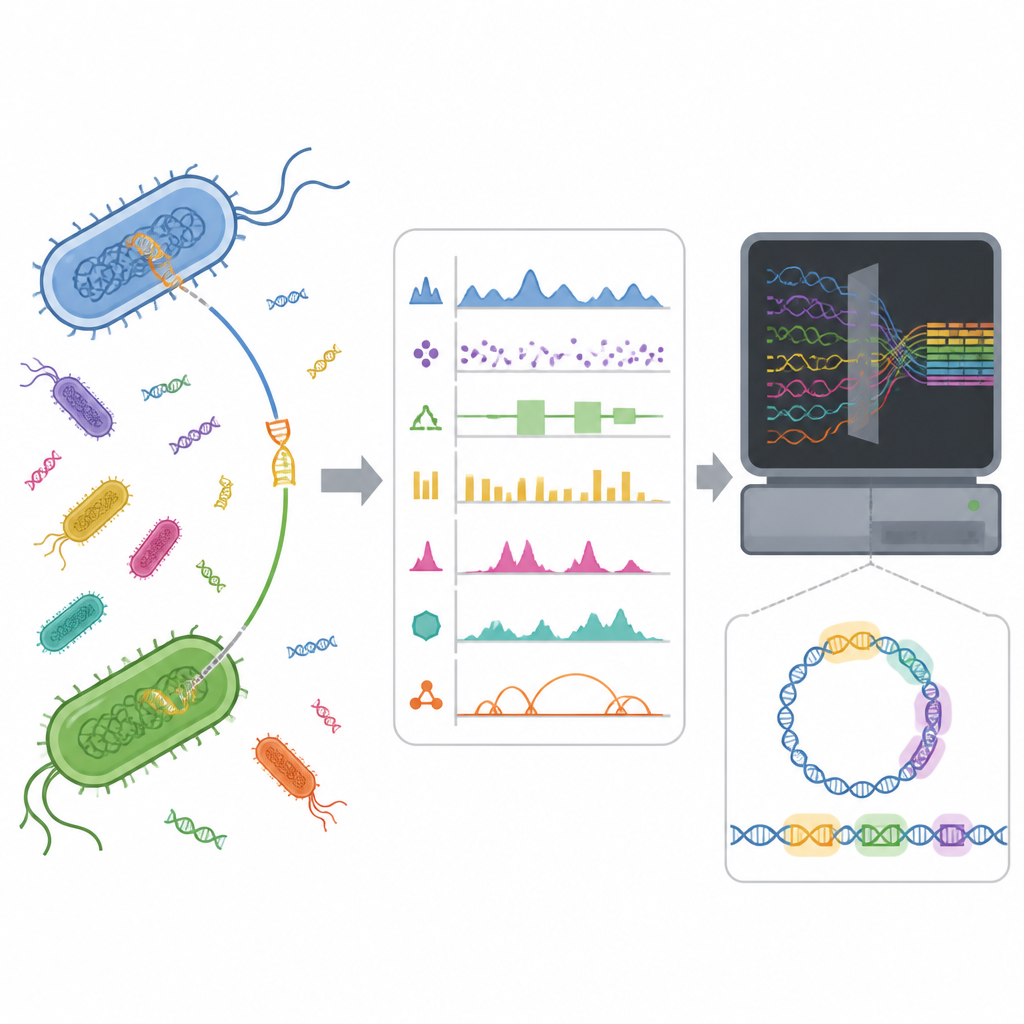
Îlots d’ADN cachés dans les génomes bactériens
Les bactéries ne comptent pas uniquement sur la lente accumulation de mutations sur plusieurs générations. Elles acquièrent souvent des paquets génétiques prêts à l’emploi provenant d’autres microbes via le transfert horizontal de gènes. Ces paquets, appelés îlots génomiques, peuvent porter des gènes de virulence, d’adaptation à des milieux hostiles ou de résistance aux antibiotiques. Trouver ces îlots dans un génome est difficile car ils prennent de nombreuses formes et peuvent se fondre dans l’ADN de l’hôte. Une meilleure détection peut aider les chercheurs à comprendre comment se propagent les caractères dangereux et soutenir la surveillance de la résistance aux antimicrobiens en santé publique.
Apprendre aux ordinateurs à repérer l’ADN inhabituel
Les outils informatiques cherchent à repérer les îlots génomiques en recherchant des motifs inhabituels dans la séquence d’ADN ou en comparant des génomes. Les méthodes récentes d’apprentissage automatique représentent un même segment d’ADN de plusieurs manières différentes, par exemple en comptant de courts fragments de séquence ou en résumant des propriétés chimiques. Des travaux antérieurs des auteurs ont montré que, bien qu’une représentation soit globalement la meilleure, plusieurs autres, faiblement corrélées, capturent des signaux différents mais tout aussi utiles. Cela suggérait que la combinaison de ces vues distinctes pourrait aider un modèle à reconnaître des îlots génomiques de façon plus complète que n’importe quelle vue prise isolément.
Assembler des équipes de modèles plutôt qu’un expert unique
Les chercheurs ont testé cette idée en créant un ensemble de modèles entraînés sur 44 représentations d’ADN différentes en utilisant cinq classifieurs courants. Ils ont d’abord sélectionné le meilleur modèle pour chaque représentation, puis utilisé un processus en deux étapes pour choisir des combinaisons à la fois précises et diverses dans leurs prédictions. Plusieurs stratégies d’ensemble ont été essayées, y compris un vote simple et une approche en empilement (stacking) plus structurée où un modèle séparé apprend à combiner les autres. Sur une collection de référence de segments d’ADN bactériens, les meilleurs ensembles ont légèrement amélioré des mesures comme le rappel, ce qui signifie qu’ils ont capturé davantage d’îlots génomiques que le meilleur modèle individuel, bien que les gains soient modestes et statistiquement faibles.
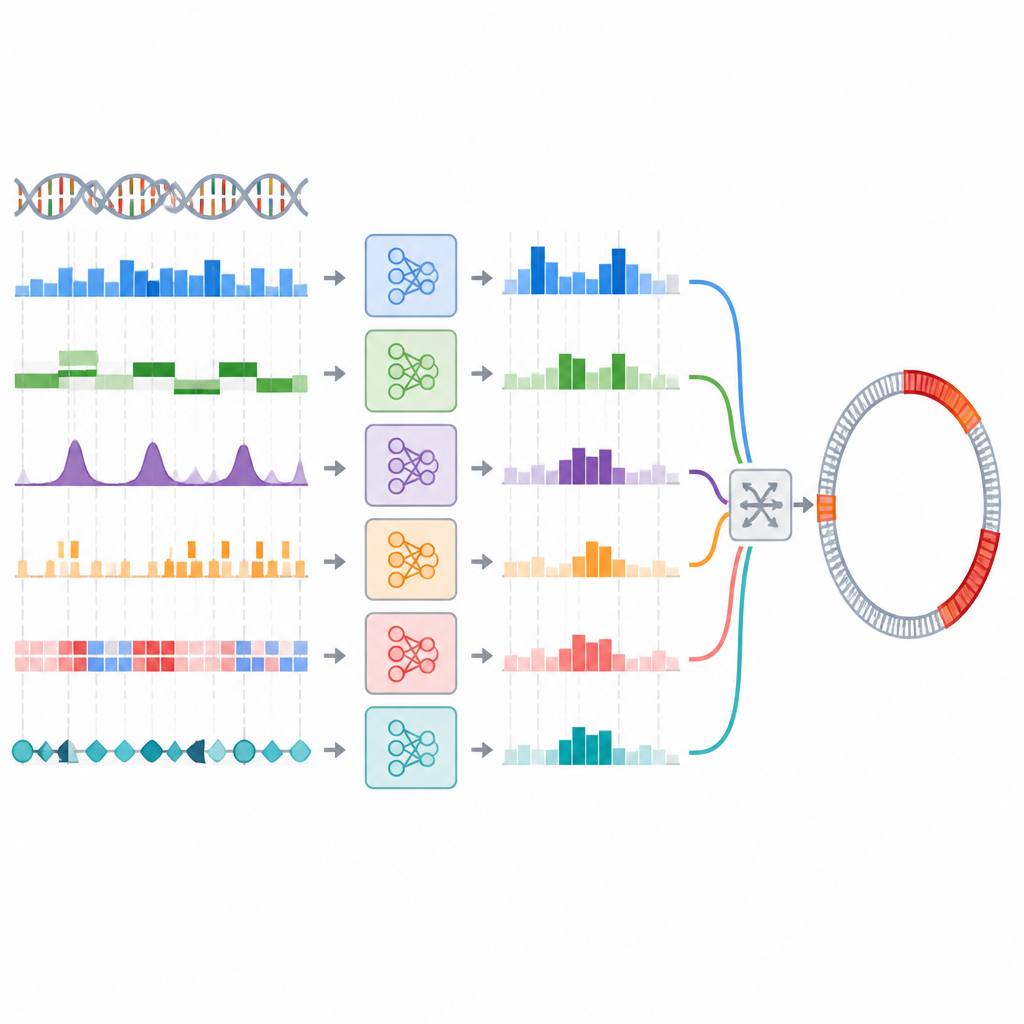
Des étiquettes de segments aux cartes réelles des génomes
En pratique, les scientifiques ont besoin non seulement d’étiqueter de courts fragments d’ADN, mais aussi de cartographier les frontières exactes des îlots génomiques le long de génomes complets. L’équipe a testé si son ensemble performant sur la tâche de classification de segments améliorerait aussi ces prédictions de frontières lorsqu’il était intégré dans un pipeline de scan de génome existant. Ici, le bilan a changé. Un ensemble basé sur le vote a peiné, manquant de nombreux îlots à moins que les seuils ne soient ajustés avec soin, et même dans ce cas n’a pas égalé le meilleur modèle individuel. Un ensemble en stacking a obtenu des performances comparables au modèle unique mais ne l’a pas clairement surpassé. Globalement, les ensembles sophistiqués n’ont pas converti leur petit avantage en classification en une meilleure cartographie à l’échelle du génome.
Repenser la formulation du problème
Les auteurs concluent que la combinaison de différentes représentations de l’ADN peut aider les modèles à repérer davantage de candidats îlots génomiques, mais que l’amélioration est limitée et sensible à la façon dont les prédictions sont utilisées. Plus important encore, l’étude montre que former des modèles uniquement pour classer des segments d’ADN prédécoupés n’est pas suffisant lorsque l’objectif réel est de dessiner des frontières d’îlots précises à travers des génomes complets. Le travail plaide pour une redéfinition de la détection d’îlots génomiques en tant que véritable problème de scan génomique, voire de régression, soutenu par de meilleurs jeux de référence et des modèles tenant compte du contexte. D’ici là, les pipelines actuels restent utiles mais doivent être appliqués avec prudence lorsqu’ils informent des études sur la propagation de la résistance aux antibiotiques.
Citation: Wijaya, A.J., Anžel, A. & Hattab, G. Evaluating ensemble learning approaches for horizontal gene transfer detection. Sci Rep 16, 16582 (2026). https://doi.org/10.1038/s41598-026-53037-x
Mots-clés: transfert horizontal de gènes, îlots génomiques, apprentissage ensembliste, résistance aux antimicrobiens, génomique et apprentissage automatique