Clear Sky Science · sv
MOFBuilder: automatiserad end-to-end-modellering av MOF-dynamik för höggenomströmmande screening
Varför små kristallkammare spelar roll
Porösa kristallina material kallade metall–organiska ramverk, eller MOF:er, är som intrikata ställningar fulla av nanoskopiska rum och korridorer. De kan fånga växthusgaser, bära läkemedel eller hysa katalysatorer, men att utforma rätt ramverk för en uppgift är extremt svårt eftersom det finns miljontals möjligheter. De flesta datorverktyg undersöker bara frusna ögonblicksbilder av dessa material, trots att deras strukturer i verkligheten flexar, andas och interagerar med vätskor och gästmolekyler. Denna artikel presenterar MOFBuilder, en mjukvarupipeline som automatiserar uppbyggnaden av realistiska, rörliga MOF-modeller så att forskare kan screena dem mycket effektivare och undvika att förbise lovande material.
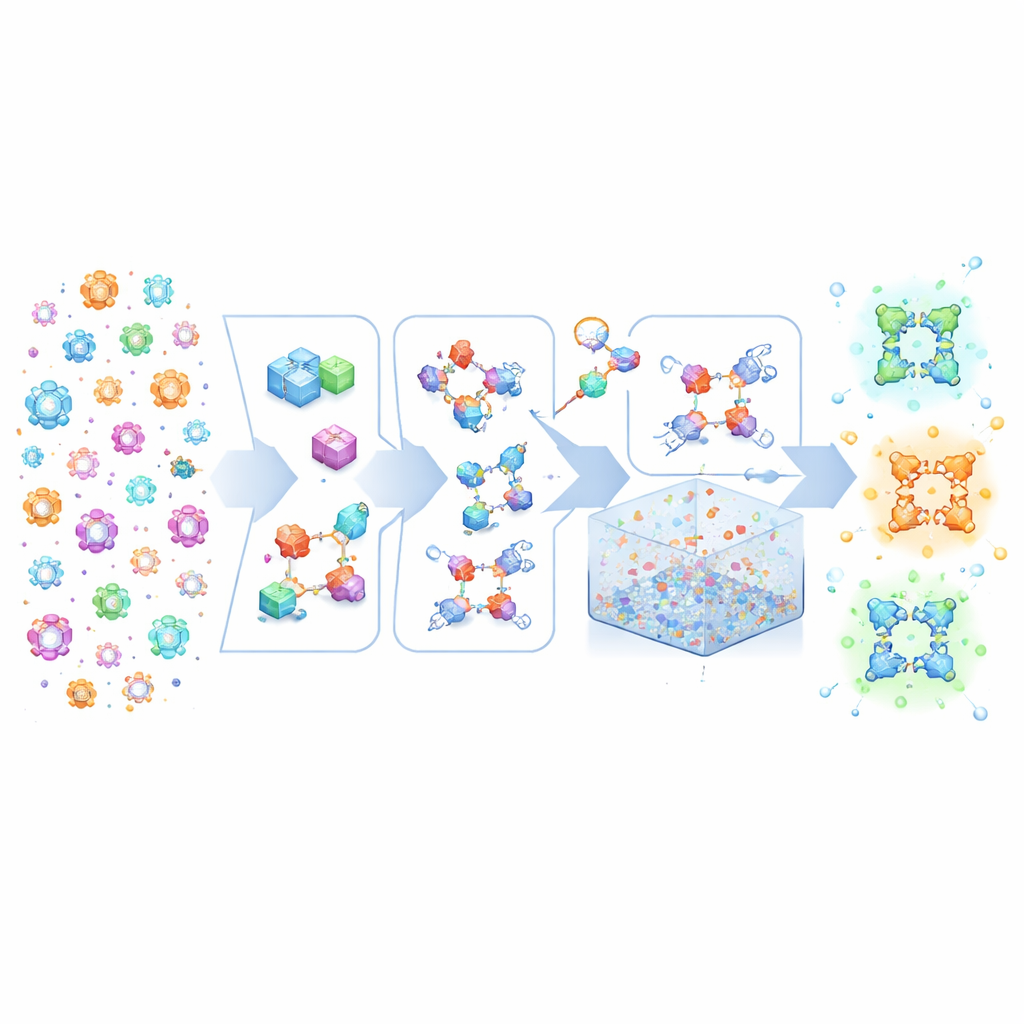
Bygga kristall-Lego från hög-nivå-recept
MOFBuilder behandlar MOF:er som om de byggdes av molekylärt Lego: metallinnehållande kluster och organiska länkar. Istället för att börja från statiska kristallografiska filer som ofta saknar fullständig kemisk detalj använder programmet en beskrivning av det övergripande nätverket plus strukturerna för dessa byggstenar. Det monterar dem sedan till ett komplett tredimensionellt ramverk samtidigt som det bevarar vilka atomer som tillhör vilken molekyl och hur de är kopplade. Detta tillvägagångssätt möjliggör generering av kemiskt konsekventa modeller på bara sekunder, inklusive idealiska oändliga kristaller, ändliga kluster, tunna skivor eller stora superceller som kan nå mikrometerskala. Eftersom modellerna bär tydliga molekylära etiketter kan de matas direkt in i vanliga molekylärdynamikmotorer utan det tidsödande manuella fixande som tidigare kunde ta veckor av expertarbete.
Redigera imperfektioner och realistiska miljöer
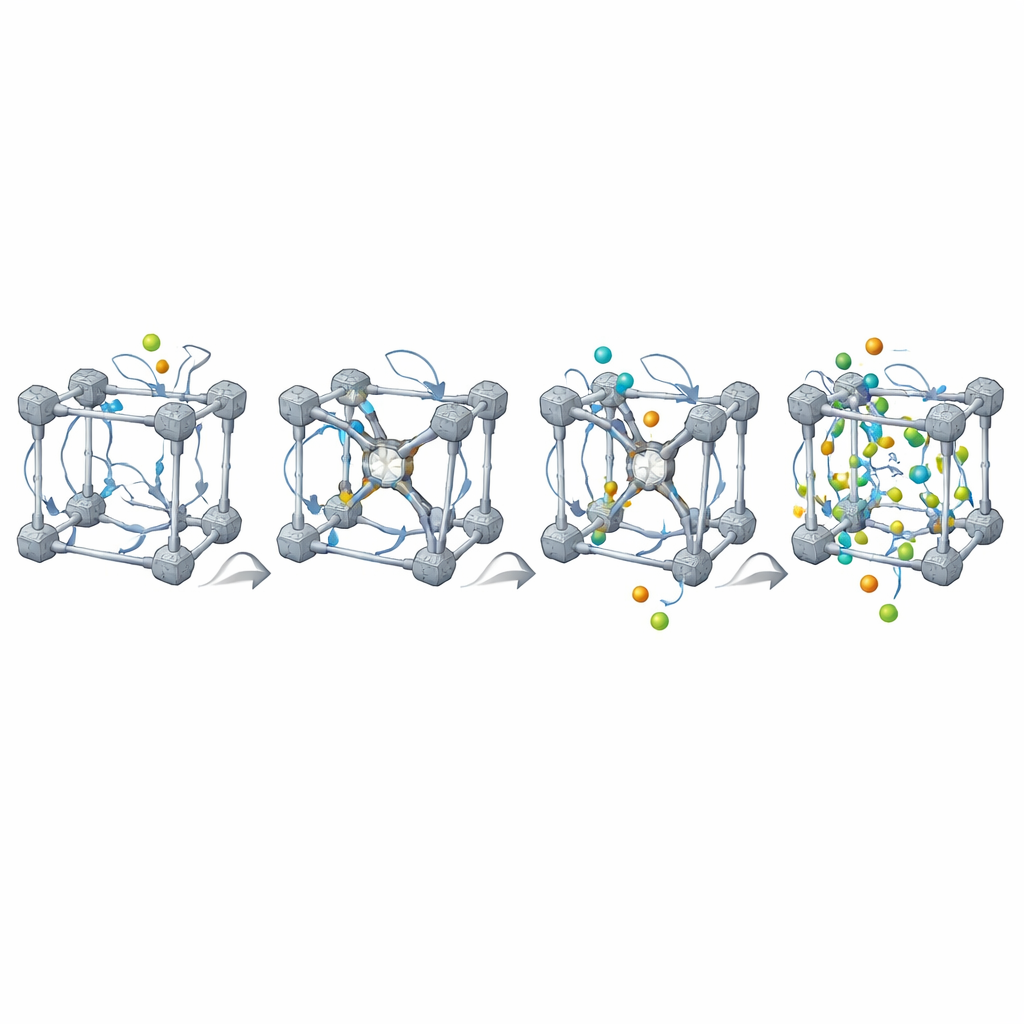
Riktiga MOF:er är aldrig perfekta: de innehåller saknade länkar och metallnoder, och ofta blandningar av olika komponenter. MOFBuilder låter användare införa dessa egenskaper direkt på nivå med byggstenarna genom att välja vilka delar som ska tas bort eller ersättas. När ett vakans skapas hittar programmet automatiskt metaller som blivit underkoordinerade och kapslar dem med små fragment så att den lokala kemin förblir rimlig och den totala laddningen balanserad. Det kan också skära ut kluster eller skivor för att studera gränssnitt och fästa lösningsmedelsmolekyler runt ramverket med en effektiv packningsalgoritm. Kraftfältsparametrar och atomladdningar för organiska länkar och metallnoder tilldelas genom inbyggda kopplingar till kvantkemi-verktyg, vilket producerar körklara indatafiler för välanvända simuleringspaket.
Se gästerna röra sig genom porerna
För att visa vad som blir möjligt med sådan automation presenterar författarna två fallstudier. I den första bygger de en stor modell av en MOF kallad NU-1000 fylld med saltvatten och en dubbelsträngad bit terapeutisk RNA. Hela uppsättningen, som tidigare skulle ha krävts veckor av manuell förberedelse, byggs, solvatiseras och jämviktas i en pipeline och simuleras sedan i över 100 nanosekunder. RNA:t driver gradvis från mitten av en bred kanal mot MOF-väggarna och sjunker ner i en bekväm position där det främst interagerar genom milda van der Waals-kontakter med de organiska länkarna. Viktigt är att RNA:t förblir strukturellt intakt och inte binder starkt till metallcentren, vilket stöder experimentella fynd att denna MOF kan skydda genetiskt material utan att skada det.
Avslöjar ett dolt porpussel
Den andra fallstudien tar sig an gasinfångning i en välkänd MOF-familj kallad UiO-66. Här genererar och simulerar författarna automatiskt 30 varianter vars länkar bär olika kemiska grupper, alla nedsänkta i vatten med löst koldioxid. Från varje simulering extraherar de dussintals deskriptorer som beskriver länkarna, poregeometrin och rörelsen hos CO2 och vatten. Maskininlärningsmodeller relaterar sedan dessa deskriptorer till hur många CO2-molekyler som faktiskt samlas nära länkarna. Ett slagkraftigt mönster framträder: i flera varianter rapporterar en standard geometrisk analys av en enskild fryst struktur noll åtkomlig porevolym, vilket normalt skulle diskvalificera dem som icke-porösa. Ändå visar de dynamiska simuleringarna att CO2 fortfarande ackumuleras i små håligheter tack vare subtila rotationer av länkarna som öppnar tillfälliga portar — en effekt som författarna kallar ”Porositetsparadoxen”.
Vad detta betyder för materialupptäckt
För icke-specialister är huvudbudskapet att lovande material kan felaktigt avfärdas om vi bara bedömer dem efter stela strukturbilder. MOFBuilder erbjuder ett sätt att rutinmässigt inkludera rörelse, defekter och realistiska miljöer i virtuell screening samtidigt som processen hålls snabb och automatisk. Genom att omvandla hög-nivå-designidéer till kemiskt trogna, dynamiska modeller som kan analyseras och utvinnas med maskininlärning lägger ramverket grunden för mer tillförlitlig, datadriven upptäckt av MOF:er för uppgifter som gasinfångning, katalys och läkemedelsleverans. I praktiken uppgraderar det materialmodellering från statiska ögonblicksbilder till animerade filmer, vilket hjälper forskare att se och utnyttja beteenden som tidigare var dolda.
Citering: Li, C., Ahlquist, M.S.G. MOFBuilder: automated end-to-end modeling of MOF dynamics for high-throughput screening. npj Comput Mater 12, 156 (2026). https://doi.org/10.1038/s41524-026-02086-x
Nyckelord: metall-organiska ramverk, molekylär dynamik, materialsökning, gasadsorption, beräkningsbaserad materialdesign