Clear Sky Science · en
MOFBuilder: automated end-to-end modeling of MOF dynamics for high-throughput screening
Why tiny crystal cages matter
Porous crystalline materials called metal–organic frameworks, or MOFs, are like intricate scaffolds full of nanosized rooms and corridors. They can trap greenhouse gases, carry drugs, or host catalysts, but designing the right framework for a job is extremely hard because there are millions of possibilities. Most computer tools examine only frozen snapshots of these materials, even though in reality their structures flex, breathe, and interact with liquids and guest molecules. This article introduces MOFBuilder, a software pipeline that automates the construction of realistic, moving MOF models so researchers can screen them far more efficiently and avoid overlooking promising materials.
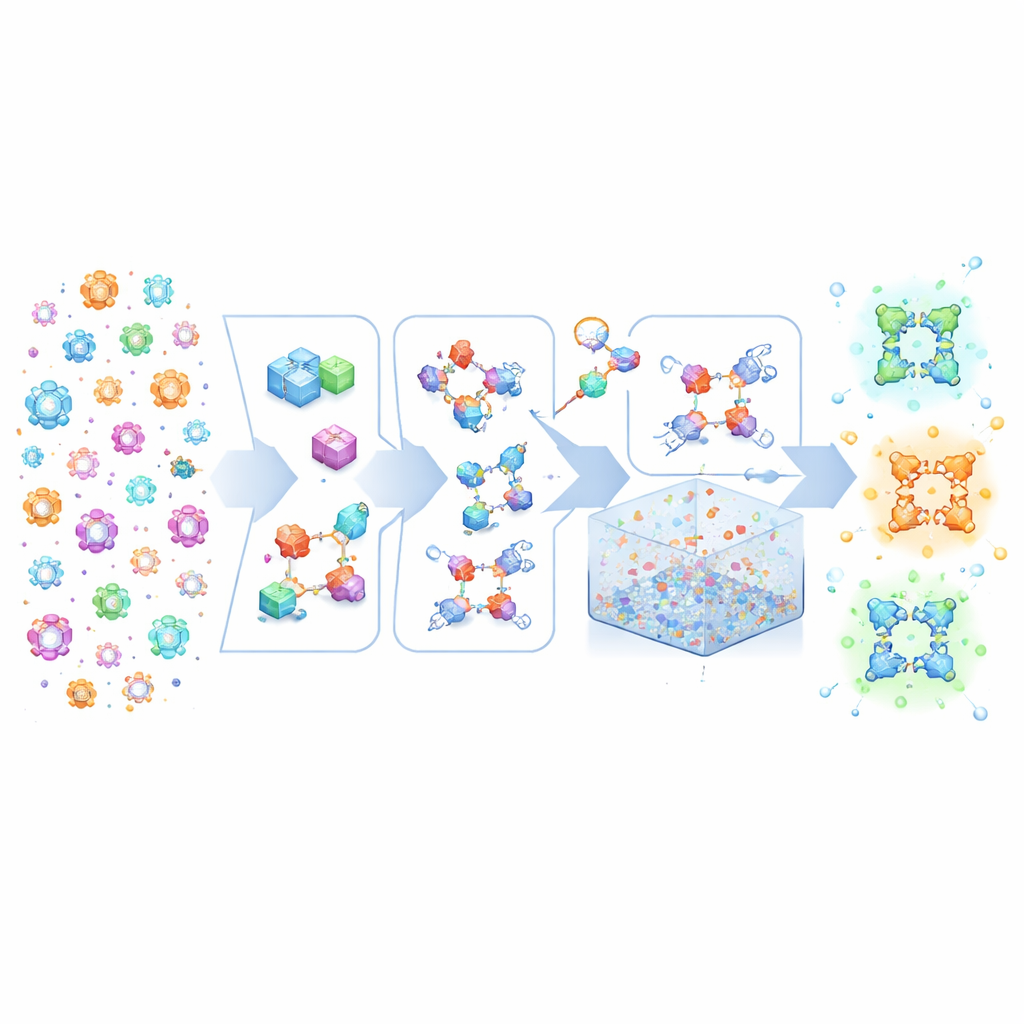
Building crystal Lego from high-level recipes
MOFBuilder treats MOFs as if they were built from molecular Lego pieces: metal-containing clusters and organic linkers. Instead of starting from static crystallographic files that often lack full chemical detail, the program uses a description of the overall network plus the structures of these building blocks. It then assembles them into a full three-dimensional framework while preserving which atoms belong to which molecule and how they are connected. This approach allows chemically consistent models to be generated in seconds, including ideal infinite crystals, finite clusters, thin slabs, or huge supercells that can reach micrometer scales. Because the models carry clean molecular labels, they can be fed directly into standard molecular dynamics engines without the laborious hand-fixing that used to consume weeks of expert time.
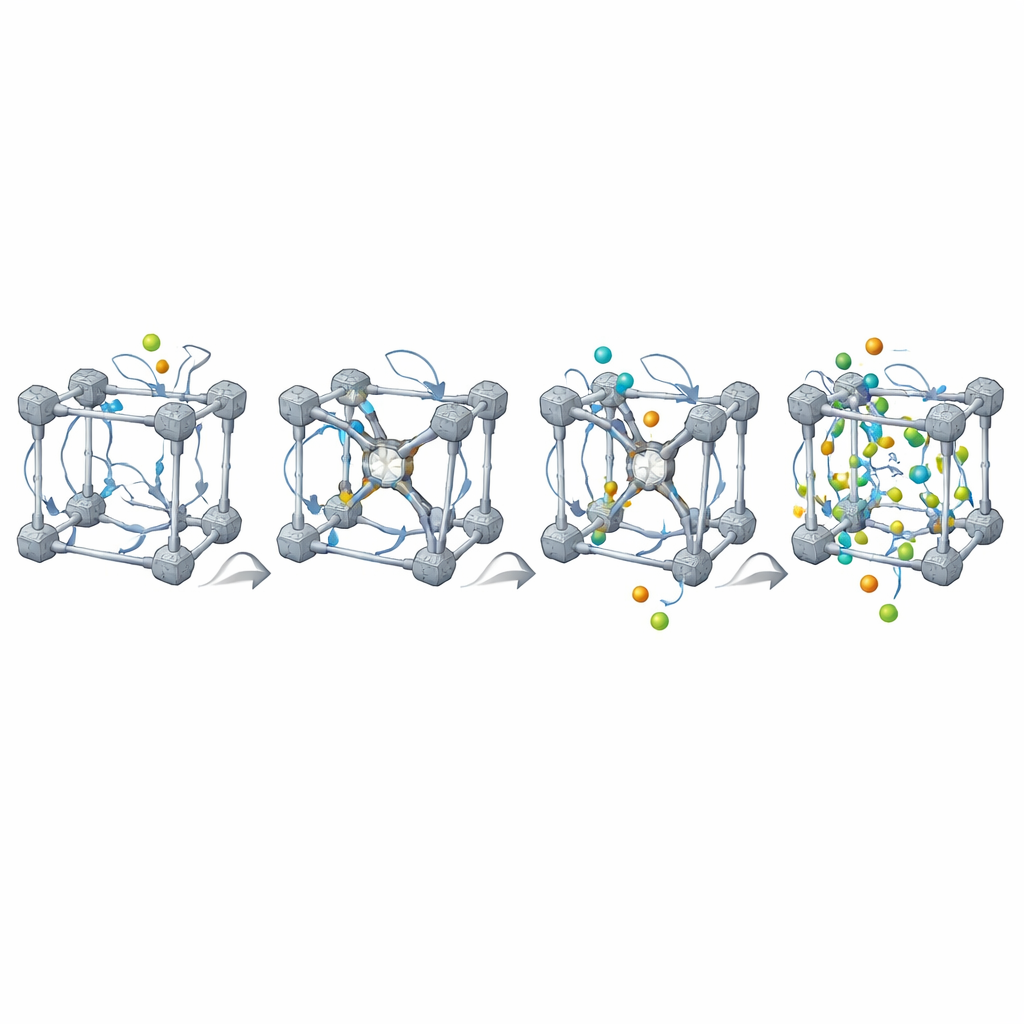
Editing imperfections and realistic environments
Real MOFs are never perfect: they contain missing linkers and metal nodes, and often mixtures of different components. MOFBuilder lets users introduce these features directly at the level of building units by selecting which pieces to remove or replace. When a vacancy is created, the program automatically finds metal sites that have become under-coordinated and caps them with small fragments so that the local chemistry remains reasonable and the overall charge stays balanced. It can also cut out clusters or slabs for studying interfaces and attach solvent molecules around the framework using an efficient packing algorithm. Force-field parameters and atomic charges for organic linkers and metal nodes are assigned through built-in links to quantum chemistry tools, producing ready-to-run input files for widely used simulation packages.
Watching guests move through the pores
To showcase what becomes possible with such automation, the authors present two case studies. In the first, they construct a large model of a MOF called NU-1000 filled with salty water and a double-stranded piece of therapeutic RNA. The whole setup, which previously would have required weeks of manual preparation, is built, solvated, and equilibrated in one pipeline and then simulated for over 100 nanoseconds. The RNA gradually drifts from the center of a broad channel toward the MOF walls, settling into a comfortable position where it interacts mainly through gentle van der Waals contacts with the organic linkers. Importantly, the RNA remains structurally intact and does not bind strongly to the metal centers, supporting experimental findings that this MOF can protect genetic material without damaging it.
Revealing a hidden pore puzzle
The second case study tackles gas capture in a well-known MOF family called UiO-66. Here, the authors automatically generate and simulate 30 variants whose linkers bear different chemical groups, all immersed in water with dissolved carbon dioxide. From each simulation they extract dozens of descriptors describing the linkers, the pore geometry, and the motion of CO2 and water. Machine-learning models then relate these descriptors to how many CO2 molecules actually gather near the linkers. A striking pattern emerges: in several variants, a standard geometric analysis of a single frozen structure reports zero accessible pore volume, which would normally disqualify them as non-porous. Yet the dynamic simulations show that CO2 still accumulates in small cavities thanks to subtle rotations of the linkers that open temporary gates—an effect the authors term the “Porosity Paradox.”
What this means for materials discovery
For non-specialists, the key message is that promising materials can be wrongly dismissed if we judge them only by rigid structural pictures. MOFBuilder provides a way to routinely include motion, defects, and realistic environments in virtual screening, while keeping the process fast and automatic. By turning high-level design ideas into chemically faithful, dynamic models that can be analyzed and mined with machine learning, the framework lays the groundwork for more reliable, data-driven discovery of MOFs for tasks such as gas capture, catalysis, and drug delivery. In essence, it upgrades materials modeling from static snapshots to animated movies, helping scientists see and exploit behavior that was previously hidden.
Citation: Li, C., Ahlquist, M.S.G. MOFBuilder: automated end-to-end modeling of MOF dynamics for high-throughput screening. npj Comput Mater 12, 156 (2026). https://doi.org/10.1038/s41524-026-02086-x
Keywords: metal-organic frameworks, molecular dynamics, materials screening, gas adsorption, computational materials design