Clear Sky Science · pl
MOFBuilder: zautomatyzowane kompleksowe modelowanie dynamiki MOF-ów do przesiewów wysokoprzepustowych
Dlaczego ważne są maleńkie kryształowe klatki
Porowate materiały krystaliczne zwane metalowo-organicznymi rusztowaniami, czyli MOF-ami, przypominają misterną konstrukcję pełną nanoskalowych komnat i korytarzy. Mogą wychwytywać gazy cieplarniane, przenosić leki lub gościć katalizatory, ale zaprojektowanie odpowiedniej struktury do konkretnego zadania jest niezwykle trudne, ponieważ możliwości jest miliony. Większość narzędzi komputerowych bada jedynie „zamrożone” migawki tych materiałów, mimo że w rzeczywistości ich struktury się wyginają, „oddychają” i wchodzą w interakcje z cieczami oraz cząsteczkami gości. W artykule przedstawiono MOFBuilder — pipeline programowy, który automatyzuje budowę realistycznych, ruchomych modeli MOF-ów, dzięki czemu badacze mogą przeprowadzać przesiewy znacznie wydajniej i nie przegapiać obiecujących materiałów.
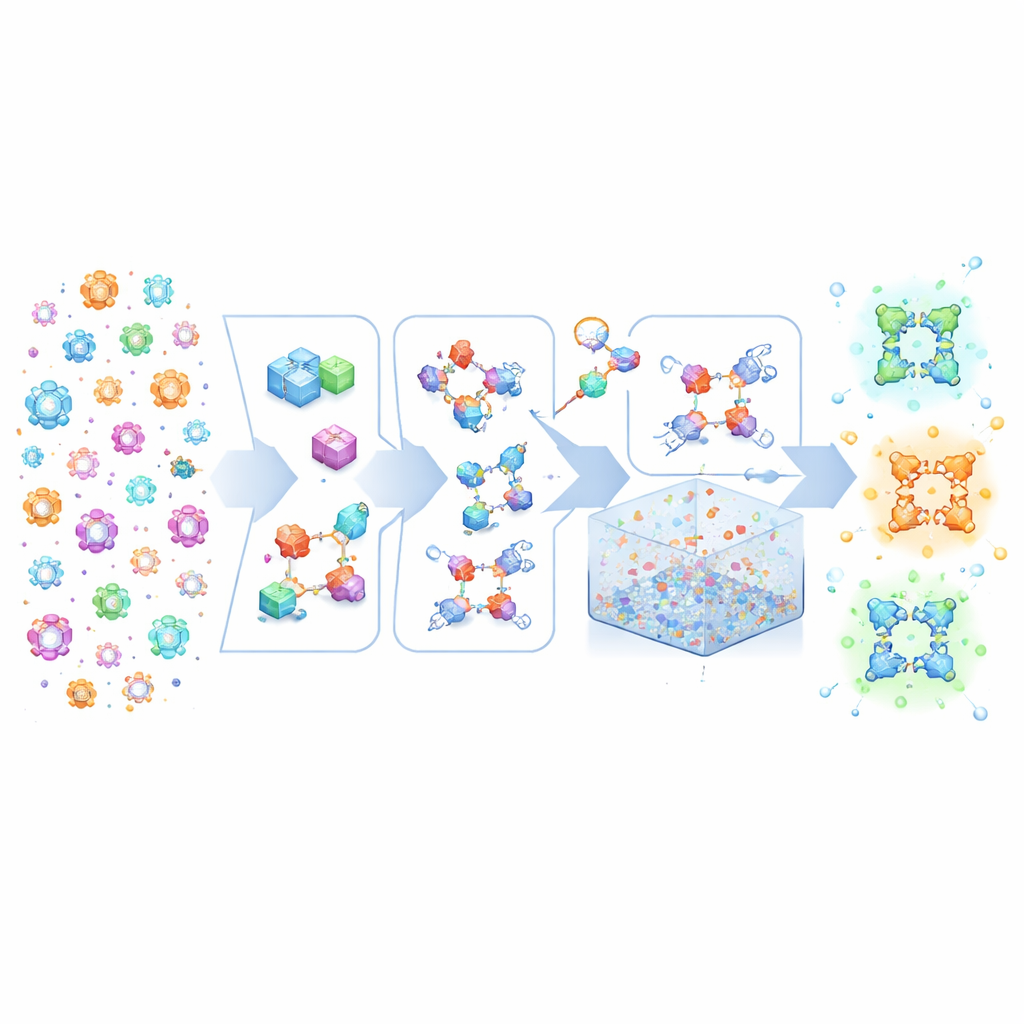
Składanie kryształowego Lego według wysokopoziomowych przepisów
MOFBuilder traktuje MOF-y jakby były zbudowane z molekularnych klocków Lego: klastrów zawierających metal oraz organicznych łączników. Zamiast rozpoczynać od statycznych plików krystalograficznych, które często nie zawierają pełnych informacji chemicznych, program wykorzystuje opis ogólnej sieci oraz struktury tych bloków budulcowych. Następnie składa je w pełne trójwymiarowe rusztowanie, zachowując przynależność atomów do poszczególnych cząsteczek i ich połączenia. Takie podejście pozwala w ciągu sekund generować chemicznie spójne modele, w tym idealne kryształy nieskończone, skończone klastry, cienkie płytki czy ogromne superkomórki sięgające skali mikrometrów. Ponieważ modele zawierają przejrzyste etykiety molekularne, można je od razu podać standardowym silnikom dynamiki molekularnej bez żmudnego ręcznego poprawiania, które dawniej zajmowało tygodnie ekspertom.
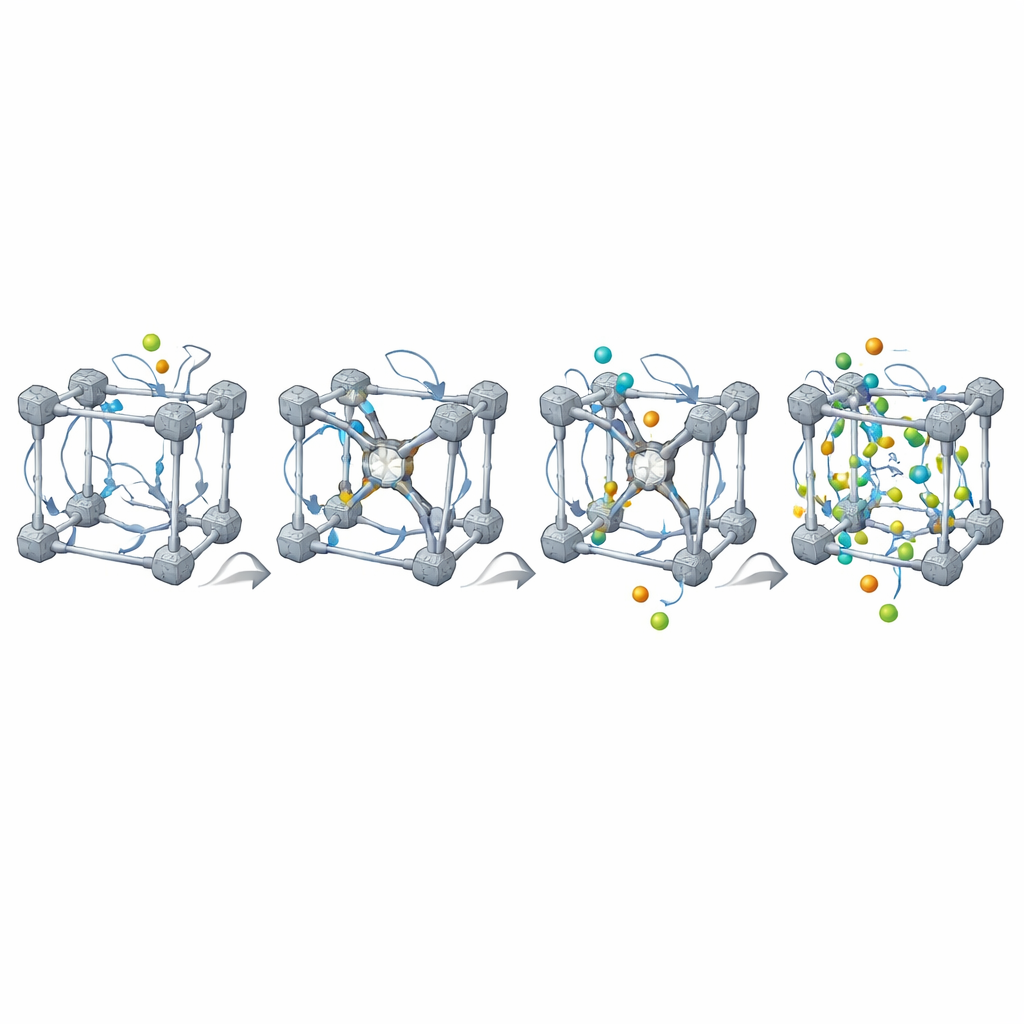
Poprawianie niedoskonałości i realistyczne otoczenia
Prawdziwe MOF-y nigdy nie są idealne: zawierają brakujące łączniki i węzły metaliczne oraz często mieszanki różnych składników. MOFBuilder pozwala użytkownikom wprowadzać takie cechy bezpośrednio na poziomie jednostek budulcowych, wybierając elementy do usunięcia lub zastąpienia. Gdy tworzy się wakans, program automatycznie znajduje miejsca metaliczne o zmniejszonej koordynacji i zabezpiecza je małymi fragmentami, tak aby lokalna chemia pozostała sensowna, a ładunek globalny był zrównoważony. Może też wycinać klastry lub płytki do badań interfejsów oraz otaczać rusztowanie cząsteczkami rozpuszczalnika przy użyciu wydajnego algorytmu upakowania. Parametry pola siłowego i ładunki atomowe dla organicznych łączników i węzłów metalicznych są przypisywane przez wbudowane połączenia z narzędziami chemii kwantowej, tworząc gotowe pliki wejściowe do powszechnie używanych pakietów symulacyjnych.
Obserwowanie ruchu gości w porach
Aby pokazać, co staje się możliwe dzięki takiej automatyzacji, autorzy przedstawiają dwa studia przypadków. W pierwszym z nich konstruują duży model MOF-u o nazwie NU-1000 wypełniony słoną wodą i podwójną nićą terapeutycznego RNA. Całe ustawienie, które wcześniej wymagałoby tygodni ręcznych przygotowań, jest zbudowane, solwatowane i wyrównane w jednym pipeline, a następnie symulowane przez ponad 100 nanosekund. RNA stopniowo przesuwa się ze środka szerokiego kanału w kierunku ścian MOF-u, osiadając w wygodnej konfiguracji, w której główną rolę odgrywają łagodne oddziaływania van der Waalsa z organicznymi łącznikami. Co istotne, RNA pozostaje strukturalnie nietknięte i nie wiąże się mocno z centrami metalicznymi, co wspiera eksperymentalne obserwacje sugerujące, że ten MOF może chronić materiał genetyczny bez jego uszkadzania.
Odkrywanie ukrytej zagadki porowatości
Drugie studium przypadku dotyczy wychwytu gazów w dobrze znanej rodzinie MOF-ów UiO-66. Autorzy automatycznie generują i symulują 30 wariantów, których łączniki noszą różne grupy chemiczne, wszystkie zanurzone w wodzie z rozpuszczonym dwutlenkiem węgla. Z każdej symulacji wydobywają dziesiątki deskryptorów opisujących łączniki, geometrię porów oraz ruch CO2 i wody. Modele uczenia maszynowego następnie korelują te deskryptory z tym, ile cząsteczek CO2 faktycznie gromadzi się w pobliżu łączników. Wyłania się uderzający wzorzec: w kilku wariantach standardowa analiza geometryczna pojedynczej zamrożonej struktury wskazuje zero dostępnej objętości porów, co normalnie dyskwalifikowałoby je jako nieporowate. Tymczasem dynamiczne symulacje pokazują, że CO2 nadal akumuluje się w małych wnękach dzięki subtelnym obrotom łączników, które otwierają tymczasowe „wrota” — efekt, który autorzy nazywają „Paradoksem Porowatości”.
Co to oznacza dla odkrywania materiałów
Dla osób niebędących specjalistami kluczowy przekaz jest taki, że obiecujące materiały mogą zostać błędnie odrzucone, jeśli ocenimy je wyłącznie na podstawie sztywnych obrazów strukturalnych. MOFBuilder daje sposób na rutynowe uwzględnianie ruchu, defektów i realistycznych środowisk w wirtualnych przesiewach, przy zachowaniu szybkości i automatyzmu. Przekształcając wysokopoziomowe pomysły projektowe w chemicznie wierne, dynamiczne modele, które można analizować i eksplorować za pomocą uczenia maszynowego, ramy te tworzą podstawy bardziej wiarygodnego, opartego na danych odkrywania MOF-ów do zadań takich jak wychwyt gazów, kataliza czy dostarczanie leków. W istocie podnoszą modelowanie materiałów z biernych migawek do animowanych sekwencji, pomagając naukowcom zobaczyć i wykorzystać zachowania wcześniej ukryte.
Cytowanie: Li, C., Ahlquist, M.S.G. MOFBuilder: automated end-to-end modeling of MOF dynamics for high-throughput screening. npj Comput Mater 12, 156 (2026). https://doi.org/10.1038/s41524-026-02086-x
Słowa kluczowe: metal-organic frameworks, molecular dynamics, materials screening, gas adsorption, computational materials design