Clear Sky Science · es
MOFBuilder: modelado automatizado de extremo a extremo de la dinámica de MOF para cribado de alto rendimiento
Por qué importan las diminutas jaulas cristalinas
Los materiales cristalinos porosos llamados marcos metal-orgánicos, o MOF, son como andamiajes intrincados llenos de habitaciones y pasillos de tamaño nanométrico. Pueden atrapar gases de efecto invernadero, transportar fármacos o alojar catalizadores, pero diseñar el marco adecuado para una tarea es extremadamente difícil porque hay millones de posibilidades. La mayoría de las herramientas informáticas examinan solo instantáneas congeladas de estos materiales, aunque en realidad sus estructuras se flexionan, respiran e interactúan con líquidos y moléculas huéspedes. Este artículo presenta MOFBuilder, una canalización de software que automatiza la construcción de modelos de MOF realistas y en movimiento para que los investigadores puedan cribarlos con mucha más eficiencia y evitar pasar por alto materiales prometedores.
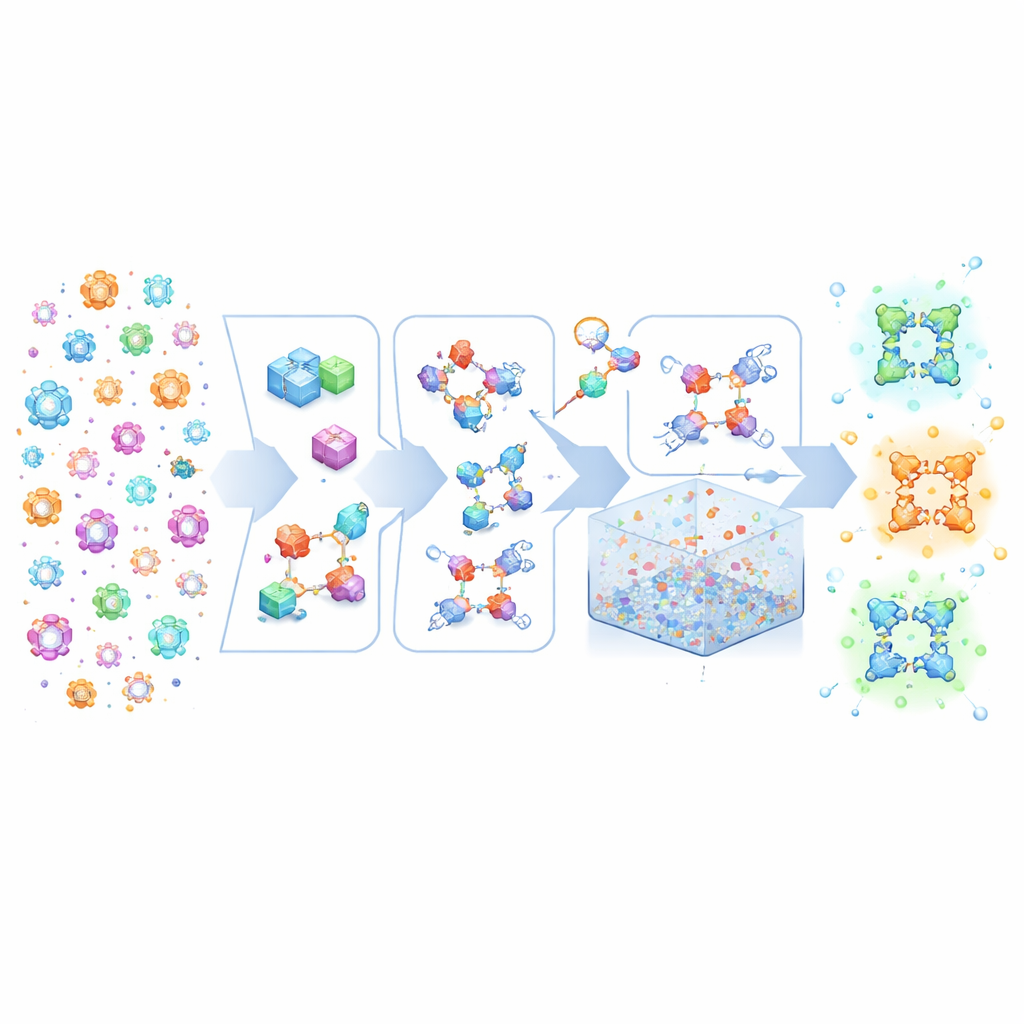
Construir Lego cristalino a partir de recetas de alto nivel
MOFBuilder trata a los MOF como si se ensamblaran con piezas tipo Lego molecular: clústeres que contienen metales y enlazadores orgánicos. En lugar de partir de archivos cristalográficos estáticos que a menudo carecen de detalle químico completo, el programa usa una descripción de la red global junto con las estructuras de estas piezas. Luego las ensambla en un marco tridimensional completo conservando qué átomos pertenecen a cada molécula y cómo están conectados. Este enfoque permite generar modelos químicamente consistentes en segundos, incluyendo cristales infinitos ideales, clústeres finitos, losas delgadas o superceldas enormes que pueden alcanzar escalas micrométricas. Como los modelos llevan etiquetas moleculares limpias, se pueden introducir directamente en motores estándar de dinámica molecular sin la laboriosa corrección manual que antes consumía semanas de tiempo experto.
Editar imperfecciones y entornos realistas
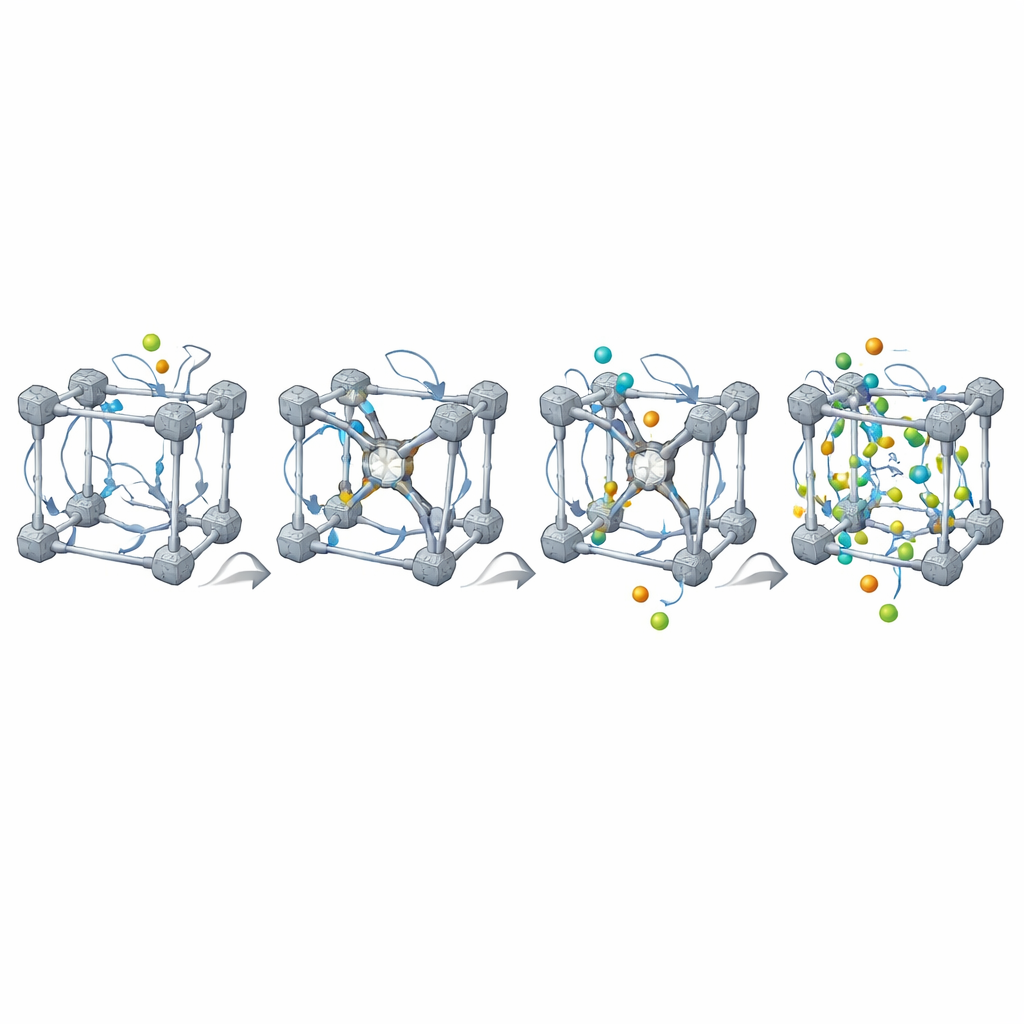
Los MOF reales nunca son perfectos: contienen enlazadores y nodos metálicos faltantes, y a menudo mezclas de distintos componentes. MOFBuilder permite a los usuarios introducir estas características directamente a nivel de unidades de construcción seleccionando qué piezas eliminar o reemplazar. Cuando se crea una vacante, el programa encuentra automáticamente los sitios metálicos que han quedado subcoordinados y los termina con pequeños fragmentos para que la química local siga siendo razonable y la carga global permanezca balanceada. También puede recortar clústeres o losas para estudiar interfaces y colocar moléculas de disolvente alrededor del marco usando un algoritmo de empaquetamiento eficiente. Los parámetros del campo de fuerza y las cargas atómicas para los enlazadores orgánicos y los nodos metálicos se asignan mediante enlaces integrados con herramientas de química cuántica, generando archivos de entrada listos para ejecutar en paquetes de simulación de uso generalizado.
Observar cómo los huéspedes se mueven por los poros
Para mostrar lo que posibilita tal automatización, los autores presentan dos estudios de caso. En el primero, construyen un gran modelo de un MOF llamado NU-1000 lleno de agua salada y un fragmento de ARN terapéutico de doble hebra. Todo el conjunto, que anteriormente habría requerido semanas de preparación manual, se construye, solva y equilibra en una sola canalización y luego se simula durante más de 100 nanosegundos. El ARN se desplaza gradualmente desde el centro de un canal ancho hacia las paredes del MOF, asentándose en una posición cómoda donde interactúa principalmente mediante suaves contactos de van der Waals con los enlazadores orgánicos. Importante: el ARN permanece estructuralmente intacto y no se une fuertemente a los centros metálicos, lo que respalda hallazgos experimentales de que este MOF puede proteger material genético sin dañarlo.
Revelando un enigma de poros oculto
El segundo estudio de caso aborda la captura de gases en una familia de MOF bien conocida llamada UiO-66. Aquí, los autores generan y simulan automáticamente 30 variantes cuyos enlazadores llevan distintos grupos químicos, todas inmersas en agua con dióxido de carbono disuelto. De cada simulación extraen decenas de descriptores que describen los enlazadores, la geometría de los poros y el movimiento del CO2 y del agua. Modelos de aprendizaje automático relacionan luego estos descriptores con cuántas moléculas de CO2 realmente se acumulan cerca de los enlazadores. Surge un patrón llamativo: en varias variantes, un análisis geométrico estándar de una sola estructura congelada informa volumen de poro accesible cero, lo que normalmente las descartaría como no porosas. Sin embargo, las simulaciones dinámicas muestran que el CO2 aún se acumula en pequeñas cavidades gracias a sutiles rotaciones de los enlazadores que abren compuertas temporales —un efecto que los autores denominan el “Paradoja de la Porosidad”.
Qué significa esto para el descubrimiento de materiales
Para los no especialistas, el mensaje clave es que materiales prometedores pueden ser erróneamente descartados si los juzgamos solo por imágenes estructurales rígidas. MOFBuilder ofrece una forma de incluir de rutina el movimiento, las defectos y entornos realistas en el cribado virtual, manteniendo el proceso rápido y automático. Al convertir ideas de diseño de alto nivel en modelos dinámicos y químicamente fieles que pueden analizarse y explotarse con aprendizaje automático, el marco sienta las bases para un descubrimiento de MOF más fiable y basado en datos para tareas como captura de gases, catálisis y administración de fármacos. En esencia, actualiza el modelado de materiales de instantáneas estáticas a películas animadas, ayudando a los científicos a ver y aprovechar comportamientos que antes estaban ocultos.
Cita: Li, C., Ahlquist, M.S.G. MOFBuilder: automated end-to-end modeling of MOF dynamics for high-throughput screening. npj Comput Mater 12, 156 (2026). https://doi.org/10.1038/s41524-026-02086-x
Palabras clave: marcos metal-orgánicos, dinámica molecular, cribado de materiales, adsorción de gases, diseño computacional de materiales