Clear Sky Science · de
MOFBuilder: automatisierte End-to-End-Modellierung der MOF-Dynamik für High-Throughput-Screening
Warum winzige Kristallkäfige wichtig sind
Poröse kristalline Materialien, sogenanntes metall‑organisches Gerüst (MOFs), gleichen komplizierten Gerüsten voller nanoskaliger Räume und Korridore. Sie können Treibhausgase binden, Wirkstoffe transportieren oder Katalysatoren aufnehmen, doch das passende Gerüst zu entwerfen ist extrem schwierig, weil es Millionen von Möglichkeiten gibt. Die meisten Computerwerkzeuge betrachten nur eingefrorene Momentaufnahmen dieser Materialien, obwohl ihre Strukturen in der Realität flexibel sind, „atmen“ und mit Flüssigkeiten sowie eingelagerten Molekülen interagieren. Dieser Artikel stellt MOFBuilder vor, eine Software‑Pipeline, die die Konstruktion realistischer, beweglicher MOF‑Modelle automatisiert, sodass Forschende diese viel effizienter durchsuchen können und vielversprechende Materialien nicht übersehen werden.
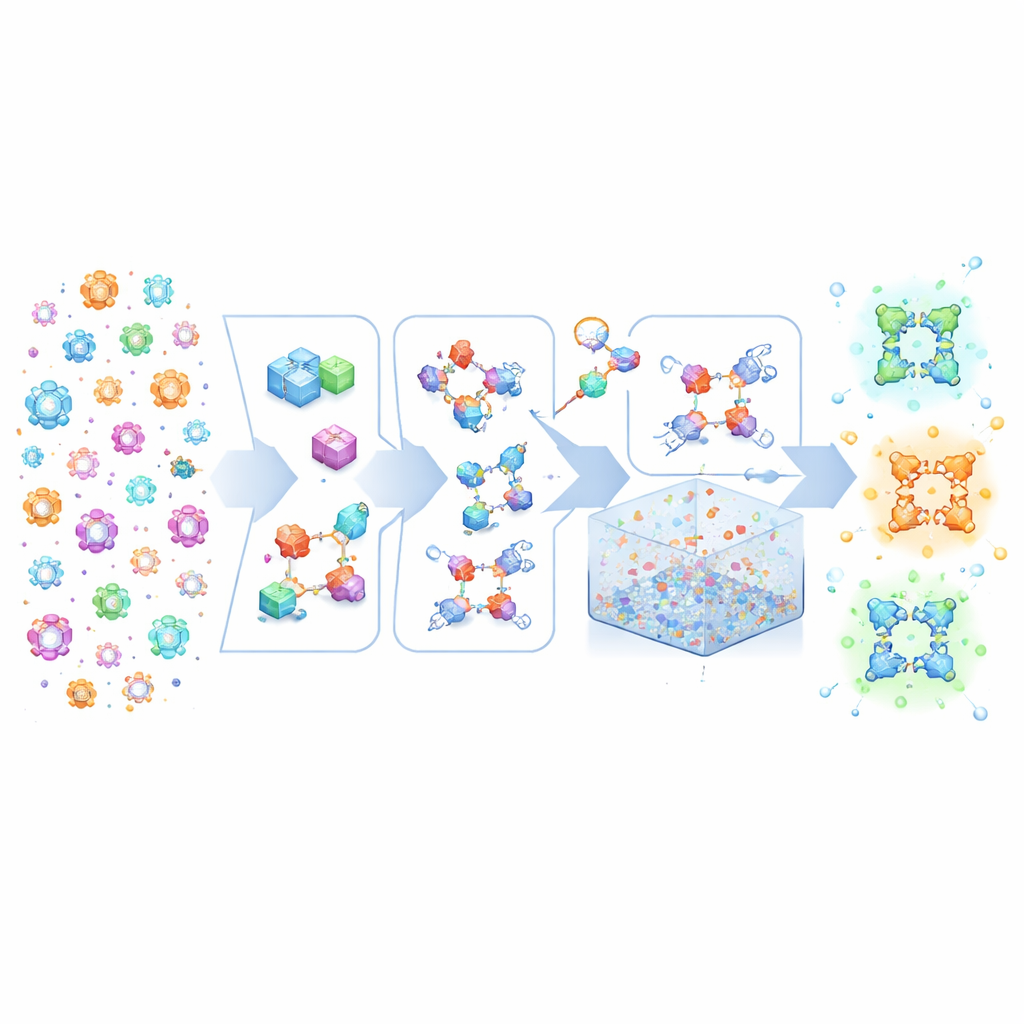
Kristall‑Lego aus hochrangigen Rezepten bauen
MOFBuilder behandelt MOFs, als wären sie aus molekularem Lego aufgebaut: metallhaltige Cluster und organische Linker. Anstatt von statischen kristallographischen Dateien auszugehen, die oft nicht alle chemischen Details enthalten, verwendet das Programm eine Beschreibung des Gesamtnetzwerks sowie die Strukturen dieser Bausteine. Es setzt sie dann zu einem vollständigen dreidimensionalen Gerüst zusammen und bewahrt dabei, welche Atome zu welchem Molekül gehören und wie sie verbunden sind. Dieser Ansatz ermöglicht die Erzeugung chemisch konsistenter Modelle in Sekunden, einschließlich idealer, unendlicher Kristalle, endlicher Cluster, dünner Schichten oder riesiger Superzellen, die Mikrometergrößen erreichen können. Da die Modelle saubere molekulare Labels tragen, lassen sie sich direkt in gängige Molekulardynamik‑Engines einspeisen, ohne die mühselige Nacharbeit, die früher Wochen Expertenzeit verschlang.
Unvollkommenheiten bearbeiten und realistische Umgebungen schaffen
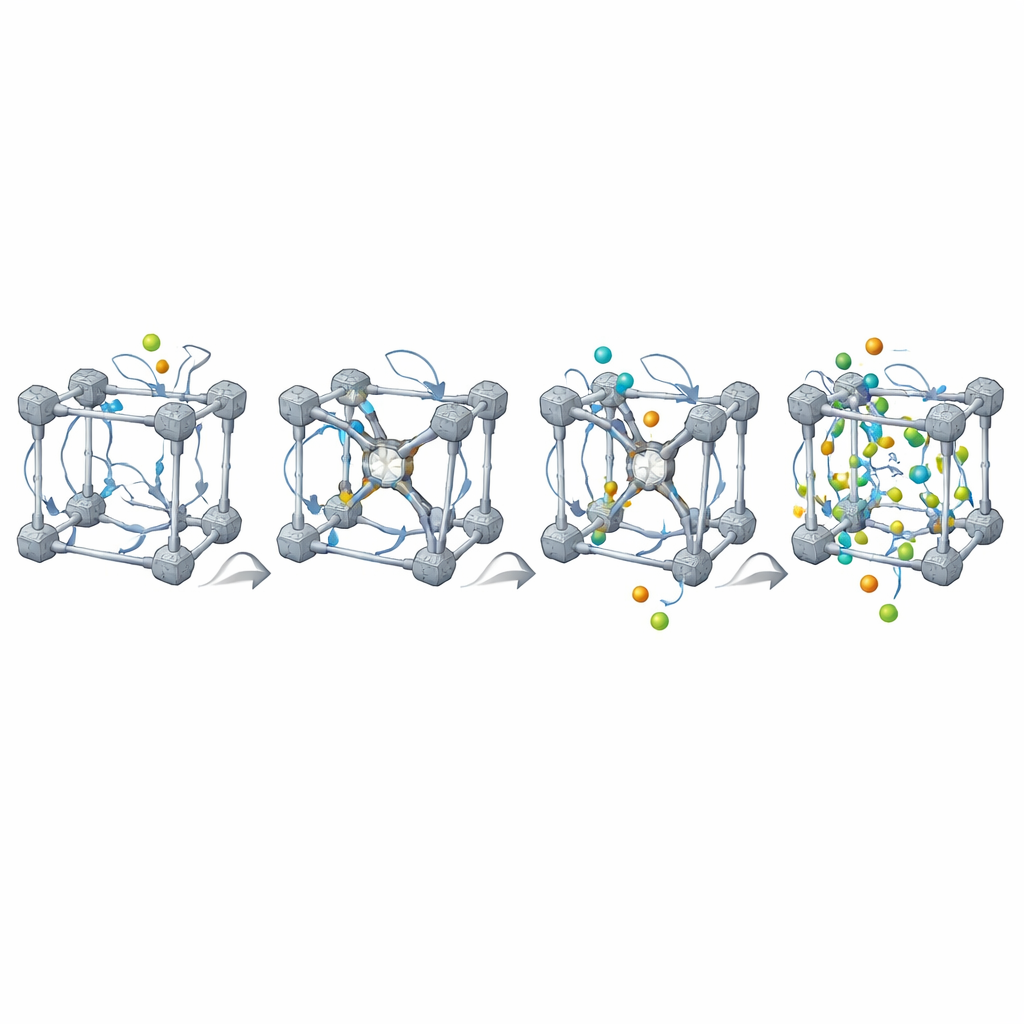
Reale MOFs sind nie perfekt: sie enthalten fehlende Linker und Metallknoten und oft Mischungen verschiedener Komponenten. MOFBuilder erlaubt es Nutzern, diese Merkmale direkt auf Ebene der Baueinheiten einzuführen, indem sie auswählen, welche Teile entfernt oder ersetzt werden sollen. Wenn eine Vakanz entsteht, findet das Programm automatisch Metallstellen, die unterkoordiniert geworden sind, und kappt sie mit kleinen Fragmenten, sodass die lokale Chemie sinnvoll bleibt und die Gesamtladung ausgeglichen ist. Es kann außerdem Cluster oder Platten ausschneiden, um Grenzflächen zu untersuchen, und Lösungsmittelmoleküle um das Gerüst herum über einen effizienten Packalgorithmus anordnen. Kraftfeldparameter und Atomladungen für organische Linker und Metallknoten werden über eingebaute Schnittstellen zu quantenchemischen Werkzeugen zugewiesen, sodass einsatzbereite Eingabedateien für weit verbreitete Simulationspakete entstehen.
Beobachten, wie Gastmoleküle durch die Poren wandern
Um zu zeigen, was mit solcher Automatisierung möglich wird, präsentieren die Autoren zwei Fallstudien. In der ersten konstruieren sie ein großes Modell eines MOFs namens NU-1000, gefüllt mit salzhaltigem Wasser und einem doppelsträngigen Stück therapeutischer RNA. Das gesamte Setup, das früher Wochen manueller Vorbereitung erfordert hätte, wird in einer Pipeline aufgebaut, solvatisiert und equilibrisiert und anschließend über mehr als 100 Nanosekunden simuliert. Die RNA driftet allmählich vom Zentrum eines breiten Kanals zu den MOF‑Wänden hin und findet eine bequeme Lage, in der sie vorwiegend durch sanfte van‑der‑Waals‑Wechselwirkungen mit den organischen Linkern interagiert. Wichtig ist, dass die RNA strukturell intakt bleibt und sich nicht stark an die Metallzentren bindet, was experimentelle Befunde stützt, dass dieser MOF genetisches Material schützen kann, ohne es zu beschädigen.
Ein verstecktes Porenrätsel aufdecken
Die zweite Fallstudie befasst sich mit Gaserfassung in einer bekannten MOF‑Familie namens UiO‑66. Hier erzeugen und simulieren die Autoren automatisch 30 Varianten, deren Linker unterschiedliche chemische Gruppen tragen, alle in Wasser mit gelöstem Kohlendioxid. Aus jeder Simulation extrahieren sie Dutzende von Deskriptoren, die die Linker, die Porengeometrie und die Bewegung von CO2 und Wasser beschreiben. Maschinelle Lernmodelle setzen diese Deskriptoren dann in Beziehung dazu, wie viele CO2‑Moleküle sich tatsächlich in der Nähe der Linker ansammeln. Ein eindrückliches Muster zeigt sich: Bei mehreren Varianten meldet eine standardmäßige geometrische Analyse einer einzigen eingefrorenen Struktur null zugängliches Porenvolumen, was sie normalerweise als nicht‑porös disqualifizieren würde. Die dynamischen Simulationen zeigen jedoch, dass sich CO2 dennoch in kleinen Hohlräumen anreichert, dank subtiler Rotationen der Linker, die temporäre Tore öffnen — ein Effekt, den die Autoren als „Porositätsparadoxon" bezeichnen.
Was das für die Materialentdeckung bedeutet
Für Nicht‑Spezialisten lautet die Kernbotschaft: Vielversprechende Materialien können fälschlich verworfen werden, wenn wir sie nur nach starren Strukturabbildungen beurteilen. MOFBuilder bietet einen Weg, Bewegung, Defekte und realistische Umgebungen routinemäßig in virtuelle Screenings einzubeziehen, dabei schnell und automatisiert zu bleiben. Indem hochrangige Designideen in chemisch getreue, dynamische Modelle umgesetzt werden, die sich mit Maschinellem Lernen analysieren und auswerten lassen, schafft die Plattform die Grundlage für zuverlässigere, datengetriebene Entdeckungen von MOFs für Aufgaben wie Gaserfassung, Katalyse und Wirkstofffreisetzung. Im Kern hebt sie die Materialmodellierung von statischen Momentaufnahmen zu animierten Filmen an und hilft Wissenschaftlern, Verhalten zu sehen und zu nutzen, das zuvor verborgen war.
Zitation: Li, C., Ahlquist, M.S.G. MOFBuilder: automated end-to-end modeling of MOF dynamics for high-throughput screening. npj Comput Mater 12, 156 (2026). https://doi.org/10.1038/s41524-026-02086-x
Schlüsselwörter: metall-organische Gerüste, molekulardynamik, Materialscreening, Gasadsorption, computationales Materialdesign