Clear Sky Science · sv
Ramverk för att helt kringgå dyra DFT-beräkningar med grafneuronät för att förutsäga vakansbildningsenergi i FCC high entropy-legeringar
Varför smartare metallutformning spelar roll
Från jetmotorer till kärnreaktorer förlitar sig ingenjörer på avancerade metalllegeringar som tål värme, påfrestning och strålning under många år. En särskild klass kallad high entropy-legeringar blandar flera metaller, vilket skapar komplexa atomära grannskap som kan göra dessa material både starka och stabila. Men att förutsäga hur små ofullkomligheter inuti dem påverkar prestanda kräver ofta extremt kostsamma kvantberäkningar. Denna studie introducerar en genväg som ersätter majoriteten av den tunga beräkningsbördan med maskininlärning, vilket öppnar dörren för snabbare utforskning av nya, tåligare legeringar. 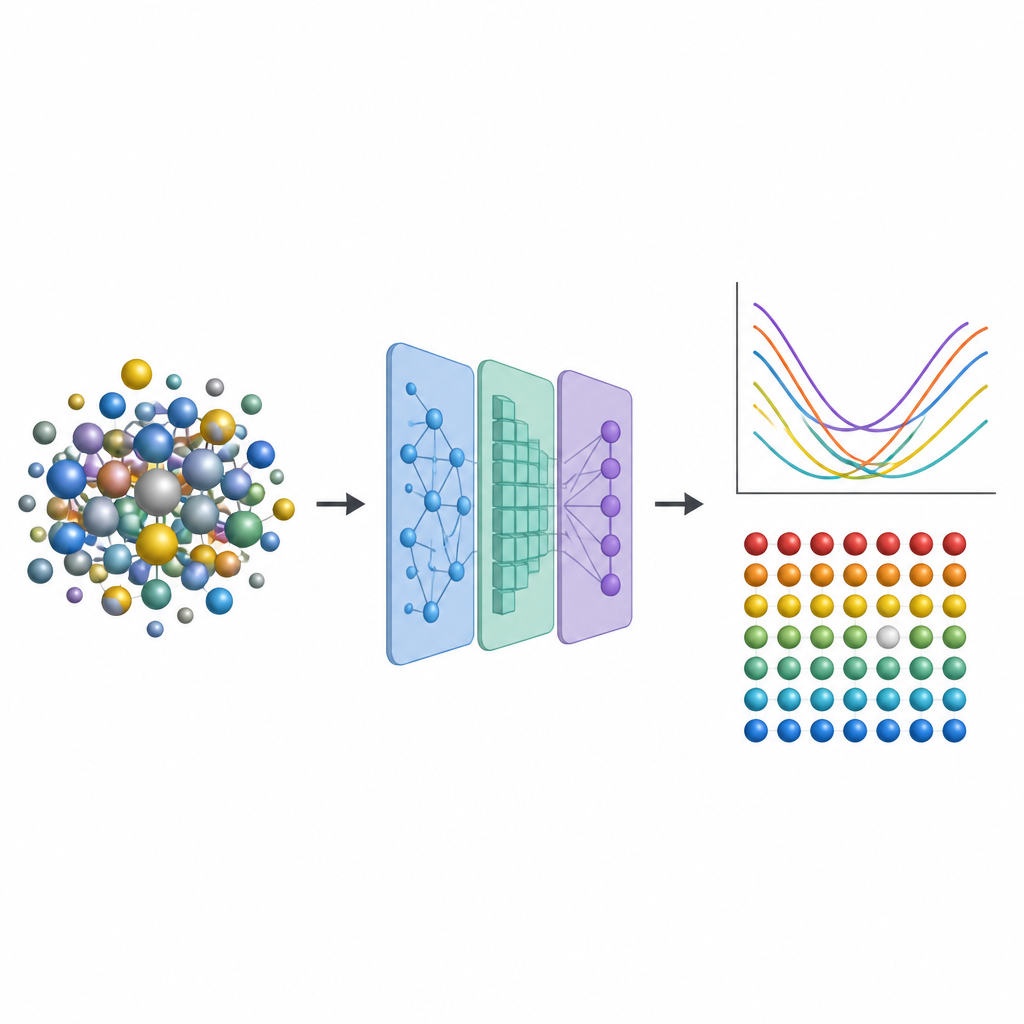
Små glapp som styr stora materialförändringar
I vilken metall som helst saknas alltid ett fåtal atomer från sina ordnade positioner och bildar vakansplatser. Dessa mikroskopiska glapp kontrollerar hur atomer rör sig, hur en metall deformeras och hur den långsamt förändras under påfrestning eller värme. Ett nyckeltal, vakansbildningsenergi, mäter hur svårt det är att skapa ett sådant glapp. I high entropy-legeringar, där många olika atomer med skilda storlekar och elektroniska tendenser blandas slumpmässigt, kan denna energi variera kraftigt från plats till plats. Att förstå den variationen är avgörande för att förutsäga kryphållfasthet, styrka och långtidstabilitet, men att beräkna den noggrant med traditionella metoder är smärtsamt långsamt.
Två enkla fingeravtryck av komplext beteende
Författarna visar att två lokala fingeravtryck fångar stor del av komplexiteten hos vakansplatser i dessa legeringar: volymen runt en atom och hur elektrisk laddning förskjuts mellan närliggande atomer. Om atomer av mycket olika storlek delar ett gitter kan utrymmet runt en potentiell vakans tänjas ut eller pressas ihop, vilket ändrar den energi som krävs för att bilda ett glapp. På samma sätt, när element med olika benägenhet att attrahera elektroner sitter intill varandra, flyter laddning mellan dem och förändrar bindningsförhållandena på ett subtilt sätt. Genom att studera enklare binära och ternära face-centered cubic-legeringar visar teamet tydliga trender: i vissa legeringar ökar vakansenergin med lokal volym, i andra följer den mängden laddning som tas upp eller avgås, och i vissa fall samspelar båda effekterna.
Bygga en digital ersättare för tunga beräkningar
För att fånga dessa effekter utan att ständigt återgå till kostsamma kvantsimuleringar utformar forskarna en trestegs maskininlärningspipeline. Först finjusterar de en befintlig neuronnätsmodell kallad CHGNet så att den kan relaxera atompositioner i slumpmässiga legeringar och reproducera de subtila gitterdeformationerna som ses i high entropy-system. För det andra tränar de ett modifierat grafneuronät för att förutsäga hur laddningen fördelas mellan atomer i dessa relaxerade strukturer, med endast information härledd från den lokala omgivningen. För det tredje matar de både strukturen och de förutsagda laddningarna in i ännu ett grafbaserat modell som ger vakansbildningsenergin för varje atomärt läge. Avgörande är att när dessa modeller väl är tränade på en modest databas av mindre, enklare legeringar kan de tillämpas på mycket större och mer komplexa legeringar utan att återgå till den ursprungliga, kostsamma kvantmetoden. 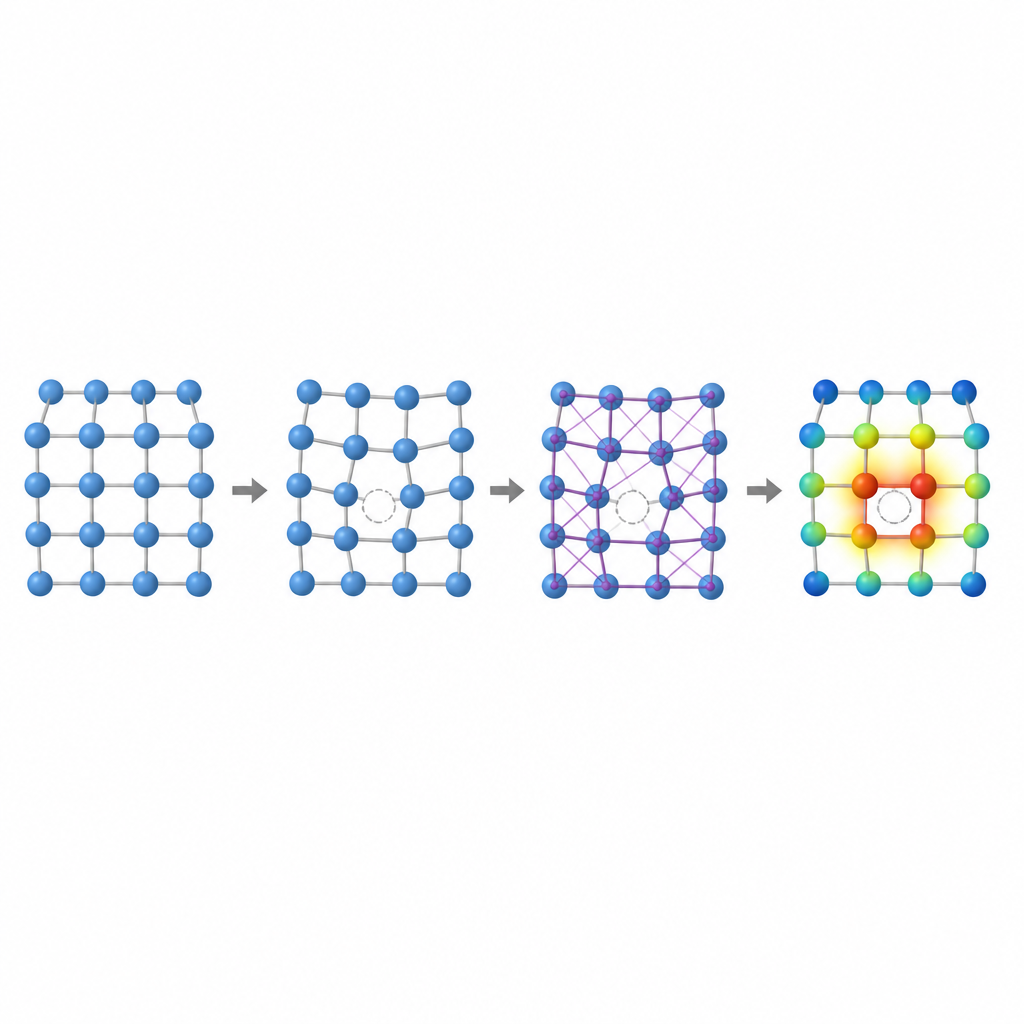
Skalning upp till realistiska legeringsstorlekar
Med detta ramverk på plats testar teamet det på nickel–koppar–guld–palladium high entropy-legeringar och vidareutvidgar sedan till legeringar som innehåller nickel, kobolt och krom. Efter en liten mängd ytterligare träningsdata från det nya systemet behåller modellerna vad de lärt sig tidigare samtidigt som de får förmågan att hantera den nya kemin. De kan förutsäga vakansenergier med en noggrannhet nära kvantberäkningarnas, men till en bråkdel av kostnaden och tiden. Metoden fungerar även på superceller med tusentals atomer, vilket ligger långt utanför räckvidden för standard kvantsimuleringar, och gör det möjligt för forskare att utforska mer realistiska representationer av oordnade legeringar.
Vad detta betyder för framtida material
Enkelt uttryckt visar detta arbete att omsorgsfullt designade maskininlärningsmodeller kan fungera som en pålitlig ersättare för tunga fysikberäkningar när man studerar små defekter i komplexa legeringar. Genom att fokusera på några få fysikaliskt meningsfulla fingeravtryck och träna på enklare system skapar författarna ett verktyg som snabbt kan skanna enorma designutrymmen för legeringar med önskvärda defektbeteenden. Detta ersätter inte behovet av kvantberäkningar helt, men det minskar kraftigt hur ofta de behövs, vilket gör det mycket mer praktiskt att söka och förfina nästa generations strukturella material.
Citering: Linton, N., Singh, P. & Aidhy, D.S. Framework to completely bypass expensive DFT calculations via graph neural networks for vacancy formation energy predictions in FCC high entropy alloys. npj Comput Mater 12, 175 (2026). https://doi.org/10.1038/s41524-026-02037-6
Nyckelord: high entropy alloys, vacancy formation energy, graph neural networks, density functional theory, machine learning in materials